调控蛋白质稳态的抗肿瘤新靶点和新策略研究进展
2022-02-09袁涛钱美佳葛孚晶杜佳泯王姣严芳洁朱虹杨波
袁涛 ,钱美佳 ,葛孚晶 ,杜佳泯 ,王姣 ,严芳洁 ,朱虹 ,杨波
(1. 浙江大学智能创新药物研究院,浙江 杭州 310058;2. 浙江省抗肿瘤药物临床前研究重点实验室,浙江 杭州 310058)
蛋白质是构成细胞的重要成分,也是维持和调控生命活动的关键物质基础。蛋白质表达、折叠、功能或定位的异常均影响机体的正常生理功能[1],因此维持这些过程处于稳定和平衡的状态,即保持蛋白质稳态,是确保生命体正常生理活动的关键[1-2]。蛋白质稳态主要由蛋白合成和降解2个过程来动态调控,其中蛋白质合成是调节蛋白质稳态的起始阶段,新合成的蛋白质经过不同种类的翻译后修饰被运输到细胞内不同部位,具有功能多样性;降解则是避免蛋白质错误折叠或异常定位与累积的关键终端调节机制,是维持蛋白质稳态的“守门员”[3]。泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)和自噬-溶酶体系统(autophagic-lysosomal system,ALS)是介导细胞内蛋白质降解的2种主要途径,这2种途径相互独立又相辅相成,能快速高效地降解功能失常的蛋白质或受损的细胞器,是调控细胞内蛋白质稳态的重要方式[4]。
蛋白质稳态不仅在维持机体正常生理功能中起到关键作用,还在应激条件下细胞命运的抉择中发挥重要作用。一旦蛋白质稳态失调,就有可能导致包括恶性肿瘤、神经退行性疾病等疾病发生。诸多研究表明,蛋白质稳态重要的调控系统包括UPS和ALS等的功能异常是致癌蛋白异常累积的重要原因[4]。此外,肿瘤在快速发生发展过程中,往往容易形成缺氧、营养耗竭的肿瘤微环境,引起肿瘤细胞内质网应激,从而激活未折叠蛋白反应和氧化应激反应,造成蛋白质稳态失调,导致肿瘤血管生成、肿瘤侵袭转移以及免疫逃逸[5]。
由此可见,蛋白质稳态异常与肿瘤演进各个阶段均有着密切的联系,是肿瘤发生发展的关键原因之一。因而,基于蛋白质稳态调控机制发展干预策略可为抗肿瘤药物的研发提供重要的新思路和新方向。本文综述了近年来蛋白质稳态调控领域的抗肿瘤新靶点和新策略的研究现状,并对其发展前景进行展望,以期为蛋白质稳态调控机制在肿瘤及相关疾病治疗中的进一步研发和应用提供参考。
1 基于调控蛋白质稳态的抗肿瘤靶点和策略
1.1 蛋白酶体及其靶向抑制剂
26S蛋白酶体是真核细胞中负责蛋白质降解的主要细胞器,由1个20S蛋白酶体核心颗粒及覆盖于一端或两端的19S调节颗粒组成(见图1A)。20S核心颗粒(相对分子质量约700 000)包含4个堆叠空心环:2个外环和2个内环,其中2个外环形成阻碍蛋白质进入内环的屏障;而2个内环决定了蛋白酶体的活性。19S调节颗粒则可以识别带有泛素化标签的底物蛋白[6]。
26S蛋白酶体是UPS的主要成分,该系统参与细胞内80%蛋白质的降解,在调节蛋白质水平与功能方面发挥着核心作用[6]。在蛋白质泛素化修饰过程中,泛素(ubquitin,Ub)首先由泛素活化酶(ubiquitin-activating enzyme,E1)活化,形成E1-Ub复合物;进而通过转酰基作用与泛素结合酶(ubquitin-conjugation enzyme,E2)结合;最后由泛素连接酶(ubiquitin-ligase,E3)介导底物泛素化[7]。泛素化的底物蛋白由19S调节颗粒识别并回收泛素分子,随后将蛋白质解折叠后转移至20S核心颗粒,由活性蛋白酶亚基切割成肽产物,完成蛋白质降解(见图1A)。
通过介导蛋白质的降解,蛋白酶体广泛参与细胞内多种信号通路和分子过程,如:细胞周期调节、核 因 子-κB(nuclear factor-κB,NF-κB)的 激活等,在高度依赖这些通路和蛋白的恶性肿瘤细胞中,抑制蛋白酶体的功能可发挥有效的抗肿瘤作用。研究表明,在淋巴系统恶性肿瘤中,NF-κB信号往往处于异常激活的状态,蛋白酶体抑制剂硼替佐米可通过阻断NF-κB抑制蛋白(inhibitor of NFκB,IκB)的降解,进而发挥抗淋巴系统肿瘤的作用[8];然而也有研究指出,在多发性骨髓瘤(multiple myeloma,MM)中硼替佐米则可通过诱导IκBβ和受体结合蛋白2的磷酸化进而诱导NF-κB活化[9],这表明硼替佐米在不同类型肿瘤中发挥抗肿瘤作用机制有所不同。目前,蛋白酶体抑制剂已成功上市用于MM等恶性肿瘤的治疗,近年来,其在实体肿瘤中抗肿瘤作用也逐渐被挖掘。最新研究表明,人第10号染色体缺失的磷酸酶及张力蛋白同源基因(phosphatase and tensin homolog deleted on chromosome ten,PTEN)缺失的胆管癌细胞内蛋白酶体组分合成和组装过程被过度激活,导致这类胆管癌细胞更依赖于蛋白酶体通路,因此硼替佐米对PTEN功能缺陷的胆管癌表现出显著的抗肿瘤活性,提示基因突变背景可能会影响肿瘤对蛋白酶体靶向治疗的敏感性[10](见图1B)。
靶向蛋白酶体为临床血液系统肿瘤治疗带来极大的帮助,解决了MM与套细胞淋巴瘤患者无药可治、预后差等问题;但其逐渐凸显的耐药性和严重的剂量限制性毒性等问题都亟待解决。此外,目前蛋白酶体抑制剂的适应证范围仍较为局限,临床上主要应用于血液系统肿瘤的治疗,而实体肿瘤的特定基因背景与蛋白酶体抑制剂响应度之间的关系,有可能是蛋白酶体抑制剂在相关实体肿瘤中是否可应用于临床治疗的关键,值得深入研究。
1.2 靶向E3泛素连接酶
泛素化与去泛素化是调控蛋白稳态的2个关键过程,分别由E3泛素连接酶和去泛素化酶所介导。目前已报道的E3泛素连接酶约有650个成员,其中约40%已被证实在UPS中发挥作用。大量研究表明E3泛素连接酶的失调与人类肿瘤的发生发展密切相关,主要参与DNA损伤修复、基因表达和信号转导等方面,因此E3泛素连接酶已被认为是较为理想的肿瘤治疗靶点[11]。鉴于调控底物蛋白的多样性,E3泛素连接酶在恶性肿瘤中的作用较为复杂,既有促肿瘤的作用,也发挥着抑制肿瘤进程的作用,甚至同一种E3泛素连接酶,在不同的细胞背景下,其作用也有可能是双向的(见图1C)。
发挥促肿瘤作用的E3泛素连接酶调控降解的底物主要是抑癌蛋白。比如E3泛素连接酶小鼠双微小体2同系物(mouse double minute 2 protein,MDM2)在多种肿瘤中过度表达,主要通过介导抑癌蛋白P53的降解,促进肿瘤的发生发展[12]。阻断P53-MDM2相互作用的小分子抑制剂如nutlin-3等在抑制实体瘤和血液肿瘤中都取得了良好的效果[13-14]。E3泛素连接酶神经前体细胞表达发育下调蛋白4(neuronally expressed developmentally downregulated 4,NEDD4)是抑癌蛋白PTEN的负调节因子,NEDD4的上调增加了PTEN的降解,进一步增强了大鼠肉瘤病毒蛋白(rat sarcoma virus,RAS)驱动的肿瘤恶性表型[15]。当调控的是促癌底物蛋白时,E3泛素连接酶则具有强大的抗肿瘤作用。有研究表明,E3泛素连接酶羟脑苷脂(cereblon,CRBN)在肝、肾脏等多种人体器官中高表达。免疫调节酰亚胺类药物沙利度胺及其衍生物来那度胺通过介导CRBN和底物之间的相互作用来驱动靶向蛋白质降解,从而发挥抗肿瘤作用,也是目前临床唯一在用的E3泛素连接酶调节剂,广泛用于淋巴瘤和骨髓瘤的临床治疗[16]。
E3泛素连接酶在肿瘤中扮演的角色很大程度上取决于其所在的组织和细胞,特别是底物蛋白在相关组织、细胞中的丰度和功能,因此明确E3泛素连接酶调控底物蛋白的机制,进而揭示其在肿瘤发生发展中的作用,是将其开发为抗肿瘤靶点的重要前提。
1.3 靶向去泛素化酶
去泛素化酶主要介导底物蛋白泛素链的剪切和泛素分子的回收,从而抑制底物蛋白降解,或改变底物蛋白的功能和活性。截至目前,在人类细胞中发现约100个去泛素化酶,分为7大家族,分别为:泛素特异性蛋白、泛素羧基端水解酶、卵巢癌蛋白酶、JAMM/MPN 结构域相关的金属肽酶家族、马查多-约瑟夫蛋白结构域蛋白酶、单细胞趋化蛋白诱导蛋白家族和锌指 UFM1 特异性多肽蛋白酶。去泛素化酶在肿瘤细胞中往往呈现表达和活性异常的状态,由于其发挥酶活催化作用的结构域较为明确,易于用小分子抑制剂调控其活性,因此近年来逐渐成为备受关注的抗肿瘤药物作用靶点(见图1D)。自2014年以来,已有靶向29种去泛素化酶的约61个去泛素化酶小分子抑制剂被相继报道[17]。
1.3.1 细胞增殖和转移相关的去泛素化酶增殖和转移相关信号通路的异常激活是肿瘤细胞的重要特征之一。大量研究表明,去泛素化酶在肿瘤细胞的增殖和转移中发挥关键作用。河马信号通路(Hippo pathway)广泛参与了包括肝细胞肝癌、胆管癌等恶性肿瘤的生长、转移和侵袭[18],其中Yes相关蛋白(Yes-associated protein,YAP)和具有 PDZ 结合基序的转录共激活因子(transcriptional co-activator with PDZ-binding motif,TAZ)是该信号通路最为重要的下游效应蛋白。去泛素化酶USP10,USP9X以及JOSD2等通过正性调控YAP/TAZ蛋白的去泛素化过程,增加它们的蛋白稳定性和功能,发挥促肿瘤作用;敲低或抑制这些去泛素化酶,则发挥显著的抗肿瘤作用[19-21]。此外,USP9X和USP10等还可去泛素化调控转化生长因子-β(transforming growth factor-β,TGF-β)通 路 的 重 要 成 员 蛋 白SMAD4,发挥促肿瘤转移的作用,而spautin-1等USP10抑制剂可抑制肿瘤细胞的体外迁移运动[22]。
1.3.2 细胞凋亡相关的去泛素化酶逃避细胞凋亡是肿瘤细胞的基本特征之一。近年来,已发现多种去泛素化酶在调节肿瘤细胞的凋亡过程中起关键作用,通过靶向去泛素化酶促进肿瘤细胞的凋亡是一种具有前景的抗肿瘤策略。USP2通过介导细胞 FLICE 样抑制蛋白(cellular FLICE-like inhibitory protein,cFLIP)的去泛素化抑制其降解,从而抑制caspase-8二聚体的形成和激活,并最终抑制细胞凋亡过程[23]。 ML364可以通过靶向抑制USP2的去泛素化酶活功能进而诱导底物蛋白G1/S-特异性周期蛋白-D1(cyclin D1)的泛素化降解从而抑制结直肠癌和套细胞淋巴瘤的细胞周期和肿瘤生长[24]。
1.3.3 DNA损伤修复相关的去泛素化酶DNA过度复制和损伤修复异常是肿瘤细胞的另一重要特征。近年来,去泛素化酶被广泛报道参与DNA损伤修复途径,因而成为相关肿瘤的干预靶点。据报道,USP1通过去泛素化调控范科尼贫血蛋白(Fanconi anemia protein,FA)和增殖细胞核抗原(proliferating cell nuclear antigen,PCNA)促进DNA修复。在USP1缺失的肿瘤中,PCNA会发生单泛素化进而促进复制叉的停滞和核基因组不稳定性[25]。
1.3.4 蛋白酶体相关的去泛素化酶硼替佐米在临床MM中的成功应用证实了蛋白酶体在肿瘤治疗中的重要作用,靶向蛋白酶体相关的去泛素化酶也成为一种具有前景的抗肿瘤策略。PSMD14,USP14和泛素C端水解酶L5(ubiquitin C-terminal hydrolase L5,UCHL5)是与蛋白酶体相关的3种去泛素化酶,其主要功能是移除底物蛋白上的多聚泛素链,促进底物蛋白进入蛋白酶体的催化核心,从而促进底物蛋白的降解[17]。VLX1570通过抑制USP14和UCHL5的酶活性,促进肿瘤细胞内泛素化蛋白的快速积累,进而发挥抗肿瘤作用[17]。VLX1570是首个进入临床试验的去泛素化酶抑制剂,但由于不确定的临床收益,该临床研究现已被暂停(临床试验编号:NCT02372240)。
1.4 蛋白水解靶向嵌合体技术
蛋白水解靶向嵌合体(proteolysis-targeting chimera,PROTAC)是一种利用UPS对靶蛋白进行降解的技术。PROTAC通过特殊设计的“Linker”结构,将E3泛素连接酶配体和靶蛋白配体连接起来,形成“PROTAC”三联体的活性形式[26](见图1E)。由于基于多肽为配体的PROTAC活性很低,相对分子质量较大的肽类配体会大大降低PROTAC入胞能力,且多肽分子可能会产生免疫原性,因而限制了其在临床应用[27]。而基于小分子配体的PROTAC成药性大大提升,人们发现沙利度胺作为CRBN的配体,亲和力很强,非常适合作为E3泛素连接酶的配体分子,据此开发了一系列可结合CRBN的小分子,产生了CRBN-based PROTAC分子[28]。此外,研究人员又基于E3泛素连接酶希佩尔-林道肿瘤抑制因子(von Hippel-Lindau tumor suppressor,VHL)开发了更多类似理化性质的配体分子,设计出了VHL-based PROTAC分子,大幅度提高了小分子的透膜性,在细胞水平的抑制活性可以达到纳摩尔级别,为后续基于受体相互作用丝氨酸苏氨酸激酶2/雌激素相关受体α (receptor interacting serine threonine kinase 2/estrogen related receptor alpha,RIPK2/ERRα)和溴结构域蛋白4(bromodomain containing 4,BRD4)的PROTAC药物研究奠定了基础[29]。
此外,基于抗体的PROTAC(antibody-based PROTAC,AbTAC)也是一种前景良好的蛋白靶向降解策略。AbTAC本质是一种完全重组的双特异性免疫球蛋白G,抗体两端可以同时靶向细胞表面目标蛋白以及跨膜E3泛素连接酶环指蛋白43(ring finger protein 43,RNF43),进而诱导蛋白复合物细胞内化并被溶酶体降解[30](见图2B)。目前基于RNF43的AbTAC被证明可以靶向肿瘤细胞表面的程序性死亡受体配体1(programmed death-ligand 1,PD-L1),但尚不清楚RNF43是否影响目标蛋白的泛素化以及对其细胞内化的影响[31]。
PROTAC技术相对于传统小分子药物具有独特的优势。首先,PROTAC分子可以靶向降解传统意义上的“不可成药靶点”。其次,PROTAC相对于传统小分子药物具有较高的选择性。最后,PROTAC可以克服肿瘤耐药问题,例如PROTAC可有效降解突变型的B细胞受体-ABL原癌基因1(B-cell receptor-ABL proto-oncogene 1,BCR-ABL1),对T315I突变的肿瘤细胞具有较高的抑制活性[32]。当然,研究发现PROTAC也具有一定的局限性,例如相对分子质量大、生物利用度差和成药困难等,这些问题都有待进一步解决。
1.5 分子胶水
与PROTAC类似,分子胶水(molecular glues)是一类可诱导E3泛素连接酶与底物蛋白发生相互作用,促进底物蛋白经泛素化被蛋白酶体降解的小分子。与PROTAC不同的是,分子胶水可以直接介导蛋白质-蛋白质相互作用,导致靶蛋白泛素化降解[33](见图1F)。目前分子胶水研究的靶点仍然比较局限,已经进入临床的分子胶水靶点大多是偶然发现的,主要集中于:Ikaros家族锌指蛋白1/3(Ikaros family zinc finger 1/3,IKZF1/3)和G1-S相变蛋白1 (G1to S phase transition 1,GSPT1)等。首个被发现可调控蛋白稳定性的分子胶水是沙利度胺,主要原理是其结合于E3泛素连接酶CRBN蛋白,可诱导多种底物的泛素化降解。此外,芳基磺胺类药物除了用于抗菌之外,近年来还被发现可通过增强E3泛素连接酶DDB1/CUL4相关因子15(DDB1 and CUL4 associated factor 15,DCAF15)与RNA结合基序蛋白39(RNA binding motif protein 39,RBM39)的相互作用,进而诱导RBM39的泛素化降解[34]。
分子胶水能够通过蛋白降解的方式下调那些无小分子结合口袋的不可成药靶蛋白,极大地扩大了可成药靶蛋白的范围;另外,分子胶水具有化学结构简单、相对分子质量低和细胞通透性好的特点,成药性更佳。但是基于天然产物及合成的分子胶水降解剂的发现具有偶然性,缺乏合理的设计策略,限制了分子胶水发现的效率和适用性,因此基于结构导向的药物发现、计算机建模与虚拟筛选将是未来分子胶水设计的主要方向。
1.6 靶向热休克蛋白
生物体在高温等压力条件下启动的以基因表达变化为特征的细胞保护机制称为热休克反应。在此过程中,热休克蛋白(heat shock proteins,HSPs)发挥关键作用,其作为分子伴侣充当折叠酶和保持酶,协助新合成蛋白的从头折叠,并防止蛋白质聚集。细胞应激时,异常折叠的蛋白质暴露的疏水氨基酸增加,被HSP27复合物识别,随后HSP70以ATP依赖的方式置换蛋白质,使部分蛋白恢复正常折叠状态,同时将部分异常蛋白质传递给HSP90;蛋白质完成折叠后,HSP90和辅助伴侣从成熟的功能性蛋白质中解离[35](见图1G)。

图1 基于泛素-蛋白酶体系统的蛋白质稳态调控策略Figure 1 Regulation strategy of protein homeostasis based on ubiquitin-proteasome system
与正常细胞相比,肿瘤细胞对HSPs表现出较为明显的依赖性。HSPs已被报道与肿瘤细胞的增殖、转移和侵袭密切相关,在多种肿瘤中过度表达并与患者的预后不良正相关,包括肺癌、胃癌和乳腺癌等[36]。HSP90和HSP70作为热休克反应的核心伴侣分子,参与调节许多致癌蛋白如P53突变体、人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)等的表达水平和蛋白稳定性,对肿瘤发生发展具有重要作用[37-38]。HSP70和HSP27还能够通过多种途径抑制肿瘤细胞凋亡,如干预细胞凋亡蛋白表达、降低细胞溶质钙水平和稳定肿瘤细胞内的溶酶体等[39-40]。靶向抑制HSP90还能够破坏DNA修复机制,上调干扰素响应,从而增强抗肿瘤免疫反应[38]。
1.7 调控自噬过程
除了UPS之外,ALS是细胞内另一种重要的降解途径,通常与UPS等其他蛋白降解途径协同降解错误折叠或受损的蛋白质。细胞中形成的较大聚集体、半衰期长的蛋白质或与受损细胞器相关的蛋白质往往通过ALS降解(见图2A)。泛素链类型参与决定了底物蛋白的降解途径,Lys-63泛素链往往诱导底物蛋白经由ALS降解[4]。
自噬在恶性肿瘤发生发展过程中发挥双重作用。目前大多数研究认为,在肿瘤发生早期,自噬主要发挥抑癌作用,通过清除受损的蛋白质和细胞器,从而维持应激条件下的蛋白质稳态,进而抑制肿瘤发生[41]。一旦肿瘤进展到晚期,自噬则作为细胞的保护和防御机制,增强肿瘤细胞对外来刺激和压力的抵抗作用,维持肿瘤细胞的存活、代谢和生长,进而促进肿瘤的发展[42]。这一作用在特定的肿瘤类型和条件下会更为明显,如RAS通路异常激活的肿瘤表现出对自噬的高度依赖性。根据自噬过程的不同阶段,可以通过调节哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、自噬相关蛋白6(autophagy-related protein 6,ATG6)或磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)复合物活性在上游靶向自噬,也可以通过调节自噬相关蛋白4B(autophagy-related protein 4B,ATG4B)活性在下游靶向自噬。另外,还可以用溶酶体酸化抑制剂阻断自噬体与溶酶体的融合[43]。P62作为选择性自噬受体能够介导致癌蛋白的选择性自噬降解。例如,P62介导了诱导急性早幼粒细胞白血病的早幼粒白血病基因/维甲酸受体基因α(PML nuclear body scaffold/retinoic acid receptor alpha,PMLRARα)融合蛋白的选择性自噬降解,而抑制mTOR活性能够激活自噬并加速PML-RARα降解[44]。
自噬抑制剂能够与靶向治疗、放射治疗和免疫治疗联合治疗肿瘤,临床试验也已证明靶向自噬用于肿瘤治疗的安全性较为可控[45]。然而,肿瘤中的自噬途径具有高度的环境和细胞背景的依赖性,如果想要更好地调控自噬用于肿瘤治疗,需进一步了解不同条件下,自噬发生的具体过程、对肿瘤的影响以及相应的调控机制。
1.8 其他调节蛋白质稳态的策略
除了上述策略之外,尚有不少新兴的调控蛋白质稳态的抗肿瘤策略进展飞速,这些新策略正在颠覆传统的药物发现模式,为蛋白降解剂及其他相应药物的开发提供新的思路。随着对ALS研究的深入,相较于仅能降解胞内蛋白质的蛋白酶体途径,通过溶酶体进行靶向蛋白质降解的策略可有效干预细胞外及膜蛋白、受损细胞器和蛋白质聚集体。
溶酶体靶向嵌合体(lysosome-targeting chimera,LYTAC)是由与化学合成的糖肽配体融合的小分子或抗体组成,LYTAC分子可以同时结合膜蛋白或细胞外蛋白的细胞外结构域以及位于细胞表面的溶酶体靶向受体,形成三元复合物后,通过网格蛋白介导的内吞作用,导致蛋白质内化随后被降解[46](见图2B)。LYTAC可以解决PROTAC无法有效地靶向膜或细胞外蛋白的问题。目前可以被LYTAC携带的目标蛋白配体包括抗体、小分子和短肽3种类型,但由于抗体的组织通透性差,小分子配体无法靶向“不可药靶”蛋白,所以抗体和小分子在应用方面具有很大的局限性,而因为结合短肽具有易于合成、结构配比灵活等优势成为LYTAC配体的发展重点[47]。目前,有临床前研究表明,共价偶联西妥昔单抗的LYTAC分子被证明可在各种细胞系中特异性降解表皮生长因子受体(epidermal growth factor receptor,EGFR),并且偶联抗PD-L1抗体的LYTAC分子可加速细胞表面PD-L1的溶酶体降解[46]。
自噬靶向嵌合体(autophagy-targeting chimera,AUTAC)是另一种靶向蛋白降解的策略。针对细胞内无法被蛋白酶体降解的蛋白质,基于ALS的AUTAC弥补了PROTAC对于UPS过度依赖的缺陷[48]。AUTAC由靶蛋白的小分子结合剂和鸟嘌呤衍生物组成(见图2B)。鸟嘌呤衍生物引发底物蛋白K63泛素链修饰,随后被溶酶体降解。AUTAC既可降解胞质蛋白,又可通过线粒体自噬促进线粒体更新。
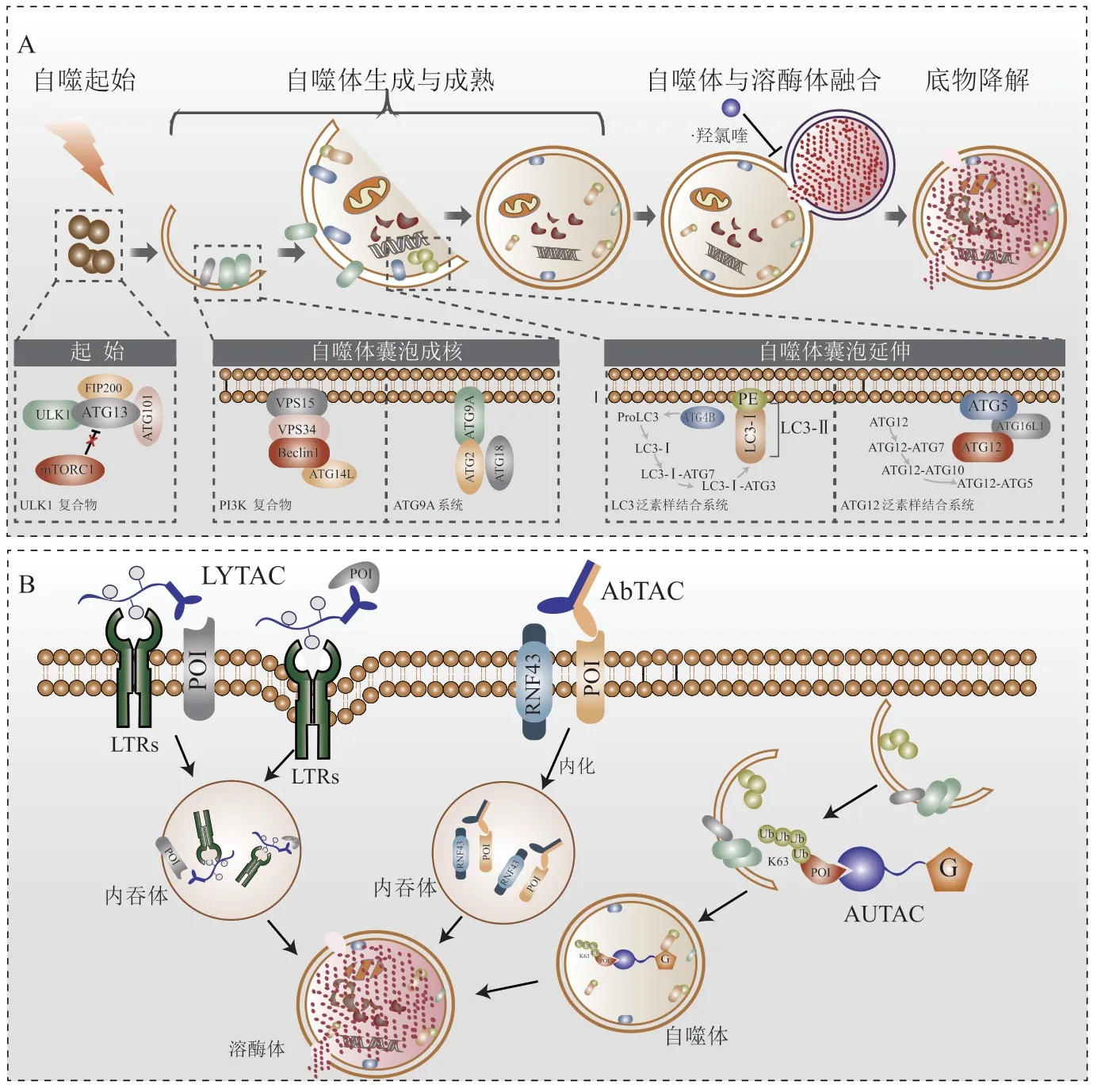
图2 基于自噬溶酶体系统的蛋白质稳态调控策略Figure 2 Regulation strategy of protein homeostasis based on autophagic-lysosomal system
2 蛋白质稳态调控药物的临床应用和临床研究 进展
2.1 已经获批上市的药物
2.1.1 蛋白酶体抑制剂近年来已成功上市了3种蛋白酶体抑制剂相关药物,即硼替佐米、卡非佐米和伊沙佐米[7]。硼替佐米是首个同类二肽硼酸盐蛋白酶体抑制剂,可逆地抑制20S蛋白酶体β5亚基的糜蛋白酶样活性,并部分抑制β1亚基的胰蛋白酶样活性;卡非佐米是第2代不可逆环氧酮蛋白酶体抑制剂,共价攻击β5亚基的活性位点苏氨酸残基;二肽基亮氨酸硼酸——伊沙佐米是首个成功开发的口服蛋白酶体抑制剂,拓宽了前两者的静脉给药途径,且具有较短的解离半衰期及良好的药动学和药效学特征。
2.1.2 E3泛素连接酶调节剂目前,唯一已上市的E3泛素连接酶调节剂是靶向调节CRBN的来那度胺及其衍生物。在上世纪50年代,沙利度胺作为孕妇止吐药广泛应用,最终酿成上万例畸形婴儿的惨剧。尽管有副作用,对沙利度胺的潜在应用仍在继续研究,并于1998年被批准用于治疗麻风病,2006年被批准用于MM。由此,揭开了沙利度胺在抗肿瘤领域中的应用,为了改善药动学和药效学性质,研究人员开发了优化的沙利度胺衍生物,如来那度胺和泊马度胺,并分别于2006年和2013年被美国食品药品监督管理局(Food and Drug Administration,FDA)批准上市。
2.2 处于临床试验阶段的候选药物及靶向策略
2.2.1 E3泛素连接酶调节剂MDM2抑制剂通过抑制MDM2的E3泛素连接酶活性并阻断MDM2和P53的相互作用发挥抗肿瘤作用。亚盛药业在研的APG-115通过抑制MDM2后激活P53,从而激活巨噬细胞 M1极化和T细胞活化的抗肿瘤免疫,也填补了我国MDM2靶点开发的空白[49]。凋亡抑制蛋白(inhibitor of apoptosis proteins,IAPs)抑制剂在促进抗肿瘤免疫中起着至关重要的作用,如LCL-161诱导MM的肿瘤坏死因子(tumor necrosis factor,TNF)依赖性凋亡,并促进抗肿瘤免疫[50]。除了开发E3泛素连接酶抑制剂外,也有激动剂在开展临床前研究。Indisulam可以充当E3泛素连接酶Cullin-4和DCAF15以及RBM39之间的分子黏合剂,在转移性乳腺癌患者中开展Ⅱ期临床研究[34](见表1)。
2.2.2 去泛素化酶抑制剂去泛素化酶明确的酶活催化位点为小分子抑制剂的开发提供了条件。该领域研究起步较晚,目前仅有VLX1570和KSQ-4279进入临床试验阶段。2015年,USP14/UCHL5选择性抑制剂VLX1570被批准用于MM患者的Ⅰ/Ⅱ期临床试验(临床试验编号:NCT02372240),然而由于临床收益不明确,目前处于暂停阶段。2017年,USP1抑制剂KSQ-4279进入Ⅰ期临床试验,用于乳腺癌易感基因(breast cancer susceptibility genes,BRCA)缺失的晚期实体瘤患者治疗(临床试验编号:NCT05240898),其机制与前述提到的USP1对DNA修复系统的调控作用有关(见表1)。
2.2.3 蛋白靶向嵌合体技术2015年,以泊马度胺为 E3泛素连接酶配体的dBET1 PROTAC成功降解了BET蛋白,PROTAC 自此进入了飞速发展期[33]。2019年,首个双功能 PROTAC 降解剂 ARV-110进入临床试验阶段,ARV-110应用于转移性去势抵抗性前列腺癌患者。另一个PROTAC分子ARV-471应用于雌激素受体(estrogen receptor,ER)阳性、HER2阴性乳腺癌患者的临床研究(临床试验编号:NCT04072952)。截至目前,已有15个PROTAC药物进入临床试验阶段(见表1)。
2.2.4 分子胶水研发进展最快的分子胶水药物目前处于Ⅱ期临床试验阶段,分别为百时美施贵宝的CC-90009,CC-92480以及C4 Therapeutics的CFT7455。CC-90009能够驱动GSPT1与CRBN结合,从而导致GSPT1的蛋白酶体依赖性降解,进而导致急性髓细胞性白血病(acute myelogenous leukemia,AML)细胞凋亡,并有效杀伤白血病干细胞,主要用于治疗复发性或难治性AML[51]。CC-92480和CFT7455都是E3泛素连接酶CRBN的调节剂,可以介导IKZF1/3的泛素化降解,主要用于治疗复发性或难治性MM[52],其中CFT7455被FDA授予MM的“孤儿药”资格认定;此外临床数据显示,CC-92480在治疗剂量下,客观缓解率可达48%(见表1)。
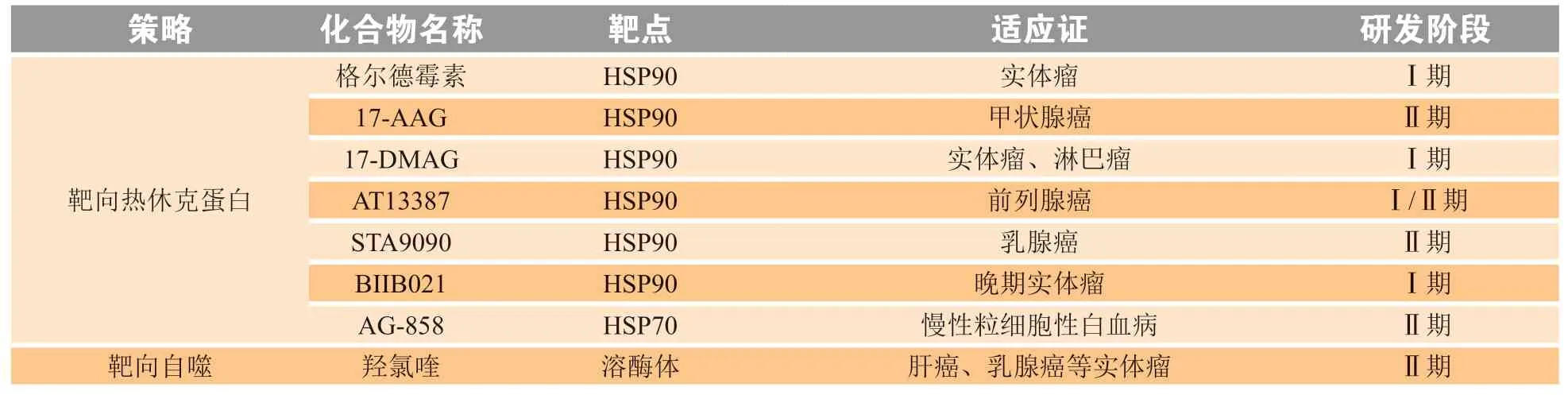
2.2.5 热休克蛋白抑制剂天然产物格尔德霉素是HSPs第1代抑制剂,其溶解性差,肝毒性大,且会通过旁路机制导致HSP70和HSP90的表达增加,引起治疗抗性。此后研究者多次进行结构改造,研发出17-AAG和17-DMAG等衍生物,一定程度上改善了毒性、稳定性等性质,但无法克服HSP70和HSP90的适应性上调[53]。第2代抑制剂AUY922,STA9090,PU-3和XL888等小分子药物较好地解决了这一问题,目前已有多个药物进入临床试验,进展最快的STA9090用于乳腺癌的治疗目前正在进行Ⅱ期临床研究(临床试验编号:NCT01273896)[53](见表1)。
2.2.6 自噬抑制剂氯喹衍生物在临床上长期用于治疗疟疾和风湿病[43]。羟氯喹能使溶酶体脱酸并阻断其与自噬体融合从而抑制自噬。目前围绕羟氯喹,科研人员正在尝试一系列联用策略,如羟氯喹与索拉非尼联用治疗肝癌,与丝裂原活化蛋白激酶(mitogen-activated protein kinase,MEK)抑制剂考比替尼及阿特珠单抗联用治疗消化道肿瘤,与MEK抑制剂曲美替尼联用治疗V-Ki-ras2 Kirsten大鼠肉瘤病毒癌基因同源物(V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog,KRAS)突变难治性胆道癌,与阿维鲁单抗及帕博西尼联用治疗乳腺癌,与达拉非尼及曲美替尼联用治疗黑色素瘤等。这些策略均处于Ⅱ期临床试验阶段(见表1)。

表1 蛋白质稳态调控药物的临床研究进展Table 1 Progress of clinical research on drugs based on proteostasis
续表1
3 结语
近年来,靶向调控蛋白质稳态的抗肿瘤策略取得了较大发展,继蛋白酶体抑制剂之后,相继有多个新品种和新靶点类型的药物用于临床治疗。围绕E3泛素连接酶、去泛素化酶在肿瘤发生发展中的作用及机制的深入研究,将进一步推动以这些蛋白质稳态调控酶为靶点的抗肿瘤药物研发进程,其中特别需要关注的是在肿瘤特定基因突变背景下异常激活并发挥重要作用的调控酶。而PROTAC,LYTAC和AUTAC等蛋白降解策略,利用UPS或ALS途径降解致癌靶蛋白,与传统靶点干预策略的“占位驱动”作用模式不同,具有独特的优势,特别是针对雄激素受体(androgen receptor,AR)的PROTAC降解剂已进入临床试验阶段,但未来研究仍需进一步关注蛋白降解策略的靶点选择及降解剂的成药性。
