儿童肿瘤分子靶向药物研究进展
2022-02-09陈一凡刘润芝王金湖徐晓军曹戟应美丹
陈一凡,刘润芝,王金湖 ,徐晓军 ,曹戟,应美丹,
(1. 浙江大学药学院,浙江 杭州 310058;2. 浙江大学医学院附属儿童医院,浙江 杭州 310003;3. 国家儿童健康与疾病临床医学研究中心,浙江 杭州 310003)
1 儿童肿瘤分子靶向药物的临床应用现状
1.1 儿童肿瘤概况
虽然近年来肿瘤的临床治疗总体上取得了重大进展,但是儿童肿瘤仍然是导致儿童非意外死亡的主要疾病之一。根据国际癌症研究机构统计,2020年全球近28万名儿童和青少年(0 ~ 19岁)被诊断患有肿瘤,近11万名儿童因肿瘤而死亡[1]。全球儿童肿瘤发病率统计显示,儿童常见的肿瘤类型包括白血病(32%)、中枢神经系统肿瘤(20%)、淋巴瘤(12%)、神经系统肿瘤(7%)等[2]。目前儿童肿瘤治疗以细胞毒类药物为主,尽管部分儿童患者因此获益,但近年来整体疗效无进一步提高。此外,细胞毒类药物的长期副作用也严重影响了儿童患者的生长发育和生活质量。最初大多数分子靶向药物是针对成人开发的,迄今为止,仅有少部分被批准用于儿童肿瘤的治疗,一些儿童特有的肿瘤类型仍面临无药可医的困境。值得注意的是,儿童肿瘤在肿瘤类型、分子特征和发病机制等方面上都与成人肿瘤有着巨大差异,这意味着针对成人肿瘤开发的靶向药物在儿童肿瘤中可能并不适用。本文系统性地总结了目前儿童肿瘤的临床药物治疗现状,回顾了目前被批准用于儿童肿瘤治疗的分子靶向药物,并在此基础上,进一步讨论基于儿童肿瘤分子特征开展靶向治疗的新策略。
1.2 儿童血液系统恶性肿瘤
1.2.1 急性淋巴细胞白血病急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)主要起源于淋巴祖细胞,是由于早期分化障碍而导致的细胞恶性增殖,是最常见的儿童肿瘤之一,在儿童肿瘤中约占24%。自20世纪中叶首次开展化疗的临床试验以来,ALL患者总生存率有了显著改善,总体生存率达90%左右[3]。尽管如此,复发的ALL儿童患者预后仍不理想,化疗后复发治愈率为50%,治疗期间复发治愈率仅20% ~ 30%[4]。
根据免疫表型不同,ALL可分为B细胞 ALL(B-ALL)和T细 胞 ALL(T-ALL),其 中 以B-ALL为主。B-ALL中常伴随染色体重排和数目异常等分子特征,这些改变往往导致融合基因的产生或基因表达失调,比如B细胞受体-ABL原癌基因1(B-cell receptor-ABL proto-oncogene 1,BCRABL1)易位、混合谱系白血病基因(mixed lineage leukemia,MLL)重排等[3]。这些基因编码许多关键的转录因子、表观遗传修饰因子或激酶,是临床上进行风险评估的主要指标。此外,组蛋白修饰基因或受体酪氨酸激酶-RAS蛋白(receptor tyrosine kinase-rat sarcoma viral oncogene homolog,RTKRAS)信号通路突变也经常在B-ALL中发生。针对具有激酶型融合或突变的B-ALL,利用激酶抑制剂进行干预是主要的策略[5]。激酶抑制剂伊马替尼(imatinib)和达沙替尼(dasatinib)靶向BCR-ABL融合蛋白中的ABL激酶结构域,在治疗具有BCRABL融合蛋白的B-ALL儿童患者中显示出较好的缓解率,为靶向儿童肿瘤中的融合基因提供了有力的证据[3]。除激酶抑制剂外,针对B-ALL的免疫靶向治疗也受到广泛关注。目前,已有3种靶向分化抗原(cluster of differentiation,CD)的药物被批准用于治疗儿童B-ALL:靶向CD19的单克隆抗体博纳吐单抗(blinatumomab)和嵌合抗原受体T细胞(chimeric antigen receptor T cell,CAR-T)疗法司利弗明(tisagenlecleucel),以及靶向CD22的抗体偶联药物奥英妥珠单抗(inotuzumab)[6-9]。
T-ALL在ALL中占10% ~ 15%[2-3]。与B-ALL相比,T-ALL的儿童患者预后较差,初次治疗后复发的T-ALL患者的无事件生存率低于25%[10]。尽管分子靶向治疗在B-ALL中取得了令人惊喜的结果,但对于T-ALL患者来说,由于缺少能够区分恶性T淋巴细胞与正常T细胞的特异性靶标,目前治疗手段仍然有限。目前,针对嘌呤类似物奈拉滨(nelarabine)和蛋白酶体抑制剂硼替佐米(bortezomib)正在开展进一步的临床试验,以确定二者是否能够改善T-ALL患者的预后[11]。
1.2.2 急性髓系白血病急性髓系白血病(acute myeloid leukemia,AML)是一类侵袭性强、异质性高的血液恶性肿瘤,在儿童肿瘤中约占6%,是预后最差的一类儿童肿瘤。尽管目前儿童AML患者的总体生存率达到约70%,但复发和难治性的AML患者预后依然较差[12-13]。此外,以细胞毒类药物为主的治疗方法还会产生长期的副作用,严重影响了儿童AML患者的正常发育和生活质量。
研究发现,一些分子信号的异常改变与AML的恶性进展、化疗耐药和复发密切相关,包括在30% ~ 35% AML患者中发生突变的FMS相关受体酪氨酸激酶3(FMS related receptor tyrosine kinase 3,FLT3),在约10% AML患者中发生突变的异柠檬 酸 脱 氢 酶(isocitrate dehydrogenase,IDH),以及在AML中异常激活的B细胞淋巴瘤2凋亡调控因子(B-cell lymphoma 2 apoptosis regulator,BCL2),Hedgehog信 号 通 路 和CD33。自2017年以来,有8种分子靶向药物被美国食品药品监督 管 理 局(Food and Drug Administration,FDA)批准用于治疗AML,包括FLT3抑制剂米哚妥林(midostaurin)和吉列替尼(gilteritinib),IDH抑制剂恩西地平(enasidenib)和艾伏尼布(ivosidenib),BCL2抑制剂维奈托克(venetoclax)、Smoothened 抑制剂以及靶向CD33的吉妥单抗(mylotarg)[14]。这些分子靶向药物多被批准用于治疗成人AML患者,在儿童AML患者中是否适用还有待进一步研究,目前仅吉妥单抗被批准用于治疗复发的或难治性的2岁以上CD33阳性的AML患者。
目前,有部分针对AML患者的分子靶点及靶向药物正处于临床试验或临床前研究阶段,展现出较好的应用前景。BCL2在细胞凋亡途径中发挥重要作用,被发现在AML中异常高表达并与AML化疗耐药密切相关[15]。临床前研究证明,BCL2抑制剂能够诱导AML细胞凋亡。目前临床试验正在评估BCL2抑制剂联合化疗在高危儿童AML患者中的治疗效果(临床试验编号:NCT03194932)[16]。
1.2.3 霍奇金淋巴瘤霍奇金淋巴瘤(Hodgkin lymphoma,HL)是B细胞来源的恶性淋巴瘤,主要发生在15 ~ 18岁青少年,其特点是肿瘤微环境中恶性细胞数量少,免疫效应细胞数量较多[17]。目前临床上HL的治疗主要以化疗或联合放化疗为主,大部分儿童HL患者预后较好,但是高危组、疾病进展以及复发的儿童患者预后仍不容乐观,此外继发性恶性肿瘤也是增加HL儿童患者预后风险的主要因素,这提示进一步寻找HL治疗新策略的重要性。
维布妥昔单抗(brentuximab vedotin)是一种抗体偶联药物,能够靶向HL细胞表面的CD30分子,经内化后释放微管抑制剂vedotin,已被批准用于治疗一线化疗失败后复发和难治性成人HL患者[18]。一项Ⅰ/Ⅱ期临床研究报告显示,维布妥昔单抗对复发和难治性儿童患者的缓解率达46%[18-19]。有文献表明程序性死亡受体1(programmed death-1,PD-1)在HL中表达异常升高,提示免疫检查点抑制剂可能在HL中有较好的响应[20]。帕姆单抗(pembrolizumab)是一种靶向PD-1的单克隆抗体,能够特异性阻断PD-1与其配体的相互作用,已在多种成人肿瘤中展现出较好的抗肿瘤活性。2017年,FDA加速批准帕姆单抗用于治疗复发和难治性HL儿童患者,临床研究表明,在15名复发和难治性HL儿童患者中有9名对帕姆单抗有较好的客观响应[21]。
1.2.4 非霍奇金淋巴瘤根据病理和组织学特征,非霍奇金淋巴瘤(non-Hodgkin lymphoma,NHL)主要分为成熟 B 细胞非霍奇金淋巴瘤(B-cell non-Hodgkin lymphoma,B-NHL)、淋巴母细胞淋巴瘤(lymphoblastic lymphoma,LBL)和间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL)。B-NHL对化疗治疗反应较好,治愈率达90%,但复发和难治性B-NHL患者预后仍然不佳[22]。目前,中高危组B-NHL患者的标准治疗采用高剂量化疗联合靶向CD20的单克隆抗体利妥昔单抗(rituximab)。CD20被认为是NHL免疫治疗的理想靶标,主要在成熟B细胞表面表达。此外,另一个针对B-NHL的研究焦点集中在CAR-T细胞上,2种靶向B细胞表面受体CD19的自体CAR-T细胞,奕凯达(Yescarta)和司利弗明(tisagenlecleucel),在B-NHL成人患者中显示出较好前景。基于此,一些临床研究正在评估该方案用于治疗复发和难治性B-NHL儿童患者的可能性[23]。
ALCL是T细胞来源的恶性淋巴瘤,占儿童NHL的10% ~ 15%[24]。研究发现,90%的ALCL儿童患者存在间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)基因易位,与肿瘤的恶性演进以及患者的不良预后相关[24]。因此,ALK抑制剂在治疗ALCL儿童患者中受到广泛关注,包括第1代ALK抑制剂克唑替尼,第2代ALK抑制剂色瑞替尼、艾乐替尼,第3代ALK抑制剂劳拉替尼等[25-26]。此外,CD30表面受体在ALCL肿瘤细胞表面表达,维布妥昔单抗是靶向CD30的抗体偶联药物,可以特异性识别CD30阳性肿瘤细胞,然后将vedotin递送到细胞中发挥细胞毒作用[18],临床研究表明维布妥昔单抗在复发性ALCL患者中显示出较好的缓解率[27]。最近一项临床试验正在研究ALK抑制剂或维布妥昔单抗联合化疗用于治疗ALCL儿童患者的效果(临床试验编号:NCT01979536)。
1.3 儿童实体肿瘤
1.3.1 神经母细胞瘤神经母细胞瘤(neuroblastoma,NB)主要由肾上腺、颈部、胸部或脊髓来源的神经母细胞发育异常形成。神经母细胞瘤通常发生于婴儿期,在0 ~ 4岁的儿童中发病率达到12.5%[28-29]。NB临床预后差异较大,低危NB患者预后较好,5年生存率可达90%。然而约60% NB患者为高危组,预后往往较差,5年生存率低于50%。对于低危组及未发生转移的NB通常采用手术切除的治疗方案,或者可能发生自发性消退。高危组患者治疗方案则主要分为3个步骤:诱导、巩固和维持。首先通过诱导化疗减小肿瘤,通过化疗和手术切除降低转移风险;然后进行高剂量化疗联合放疗,干细胞移植等巩固;后续还需采用双唾液酸神经节苷脂(disialoganglioside,GD2)单抗、粒-巨噬细胞集落刺激因子(granulocyte-macrophage colonystimulating factor,GM-CSF)和白细胞介素2(interleukin-2,IL-2)等免疫治疗或顺式维甲酸等分化治疗手段进行维持[30]。
双唾液酸神经节苷脂GD2在NB细胞上高度表达,因此被认为是一个有吸引力的治疗靶点[31]。GD2的单克隆抗体达妥西单抗(dinutuximab)和那昔妥单抗(naxitamab)已获 FDA 批准用于治疗高危NB的维持治疗,并于2021年获批进入中国市场[32-33]。ALK突变在部分NB儿童患者中发生,且ALK突变的NB儿童患者在临床上显示出更差的预后水平[34]。目前,几种ALK抑制剂进入了NB儿童患者的Ⅰ期临床试验,包括克唑替尼、色瑞替尼和恩沙替尼等[35-36]。
1.3.2 骨肿瘤骨肉瘤(osteosarcoma)是一种起源于间叶细胞的恶性骨肿瘤,随年龄呈现双峰分布特征,在10 ~ 19岁患者中发病率较高。目前临床上骨肉瘤的主要治疗策略为手术联合新辅助化疗的方法,该方法使骨肉瘤患者的5年总体生存率有了显著提高,但近年来却没有进一步改善。此外,骨肉瘤极易转移和复发,发生复发和转移的患者5年总体生存率仅为20%[37]。骨肉瘤的基因组具有高度不稳定性和复杂性,典型的分子改变为抑癌基因TP53和成视网膜细胞瘤易感基因(retinoblastoma susceptibility gene,RB)肿 瘤 抑制因子的失活。此外,α地中海贫血/智力发育迟钝综合征X相关基因(alpha thalassemia/mental retardation syndrome X-linked,ATRX),细胞周期蛋白依赖性激酶抑制剂2A(cyclin dependent kinase inhibitor 2A,CDKN2A),乳腺癌易感基因(breastcancer susceptibility gene,BRCA)和血管内皮生长因子A(vascular endothelial growth factor A,VEGFA)等基因突变,磷脂酰肌醇3-激酶-哺乳动物雷帕霉素靶点(phosphatidylinositol 3-kinasemammalian target of rapamycin,PI3K-mTOR),细胞周期等相关信号通路的改变也在骨肉瘤中较常发生[22]。围绕以上的分子特征正在开展很多临床前研究。
尤文肉瘤(Ewing sarcoma)是第二常见的骨肿瘤,特征在于尤文肉瘤RNA结合蛋白1(Ewing sarcoma RNA-binding protein 1,EWSR1)基因易位,其中最常见的是与佛氏白血病病毒整合蛋白1(Friend leukemia virus integration 1,FLI1)基因产生的融合[38]。尤文肉瘤的标准疗法以高剂量化疗为主,然而长期毒性对儿童患者具有不可逆的伤害。此外,对复发和难治性尤文肉瘤儿童患者来说,仍缺少有效的治疗手段。与骨肉瘤相比,尤文肉瘤具有较低的突变负荷,目前一些研究主要集中在EWSR1基因编码的蛋白EWS所形成的融合蛋白及其下游靶点中。最近一项研究报道,抑制EWS-FLI1融合蛋白和RNA解旋酶之间的相互作用在临床前研究中显示出较好的活性,目前正在进行Ⅰ期临床试验(临床试验编号:NCT02657005)。
1.3.3 横纹肌肉瘤横纹肌肉瘤(rhabdomyosarcoma,RMS)是儿童中最常见的软组织肉瘤,恶性程度高且预后差,目前临床上的治疗方案以手术结合放化疗为主。根据病理组织学特征,RMS可以分为腺泡型横纹肌肉瘤(alveolar rhabdomyosarcoma,ARMS)、胚胎型横纹肌肉瘤(embryonal rhabdomyosarcoma,ERMS)和多形型横纹肌肉瘤(pleomorphic rhabdomyosarcoma,PRMS)。PRMS好发于成人,ARMS与ERMS好发于5岁以上的儿童及青少年[39],其中ARMS的恶性程度最高,约80% 的ARMS患者表达配对盒基因3-叉头框O1(paired box 3-forkhead box O1,PAX3-FOXO1)融合蛋白以及PAX7-FOXO1融合蛋白,PAX3/7-FOXO1融合蛋白被认为是驱动ARMS发生及恶性进展的主要因素,其中PAX3-FOXO1融合基因阳性的ARMS患者5年生存率仅约30%[40]。
目前临床上尚无分子靶向药物被用于治疗儿童RMS患者。由于缺少可靶向的酶活口袋,目前一些研究集中在干预PAX3-FOXO1融合蛋白的稳定性或下游信号通路上,比如研究发现保罗样激酶1(Polo like kinase 1,PLK1)可通过促进PAX3-FOXO1发生磷酸化进而抑制其泛素化降解,使用PLK1抑制剂伏拉塞替(volasertib)可以在体内外有效地抑制PAX3-FOXO1阳性的ARMS[41],目前被FDA批准授予特权加快临床试验流程用于治疗横纹肌肉瘤。
1.3.4 中枢神经系统肿瘤中枢神经系统(central nervous system,CNS)肿瘤是儿童中最常见的实体肿瘤。胶质瘤是最常见的儿童CNS肿瘤,低级别胶质瘤(low-grade gliomas,LGG)儿童患者的标准治疗主要以手术切除为主,大部分患者预后较好,对于不可切除的患者通常需要进一步治疗,包括放疗和化疗。基因组测序研究显示,LGG常伴有丝裂原激活蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路相关基因,包括B-Raf原癌 基 因(B-Raf proto-oncogene,BRAF)V600E突变、神经纤维瘤蛋白1(neurofibromin 1,NF1)突变、CDKN2A突变、神经营养受体酪氨酸激酶2(neurotrophic receptor tyrosine kinase 2,NTRK2)突变或融合[42]。儿童脑肿瘤联盟研究显示使用丝裂原活化细胞外调节蛋白激酶(mitogen-activated extracellular signal-regulated kinase,MEK)抑制剂司美替尼(selumetinib)治疗复发性LGG儿童患者显示较好的效果,另一个MEK抑制剂曲美替尼(trametinib)也有类似疗效的报道[43-44]。与LGG相反,高级别胶质瘤(high-grade gliomas,HGG)往往预后较差,尽管进行了手术和辅助治疗,70% ~ 90%的儿童患者生存期不足2年[45]。目前,世界卫生组织(World Health Organization,WHO)根据组蛋白H3的突变类型进一步区分HGG亚型,每种亚型中伴随一些其他基因组变异[45]。但目前尚无分子靶向药用于HGG的临床治疗。
综上所述,分子靶向药物在儿童肿瘤的治疗上展现出良好的应用前景,不仅能延长儿童肿瘤患者的生存时间,而且相较于化疗药物具有更低的毒性,能有效改善儿童肿瘤患者的生存质量,具有巨大的临床应用价值。但是,目前分子靶向药物在儿童肿瘤中的应用覆盖面仍然十分有限,主要集中在血液肿瘤上(见表1),在实体瘤上的研究仍需加大力度探索。目前,有大量针对儿童肿瘤的分子靶向药物正处于临床试验阶段,其疗效有待明确,但也展现了儿童肿瘤分子靶向疗法发展的大趋势,将来会有越来越多的分子靶向药物可作为儿童肿瘤的临床治疗首选药物。
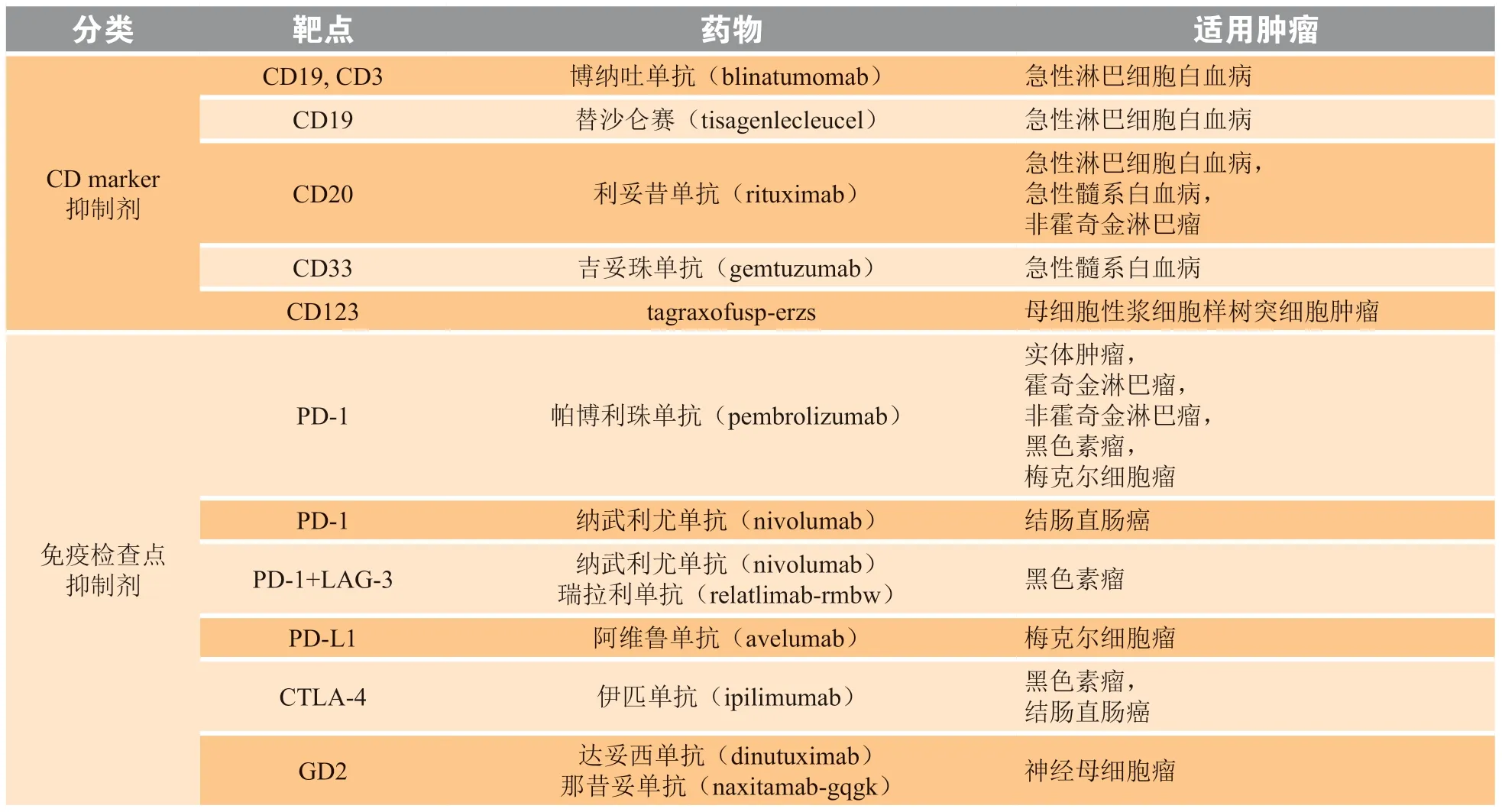
表1 被批准用于儿童肿瘤的靶向治疗药物[46]Table 1 Targeted therapies approved for pediatric tumors
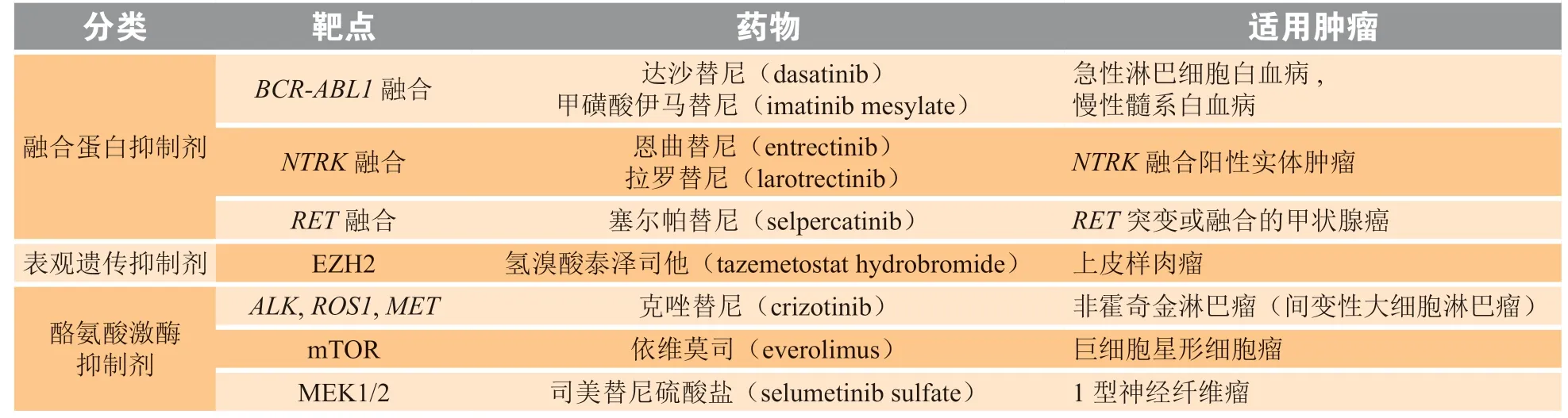
续表1
2 儿童肿瘤的主要分子特征
近年来针对儿童肿瘤的大规模基因组测序研究详细地描绘了多种儿童肿瘤的分子特征,并提供了更加有力的证据证实儿童肿瘤在起源和基因组特征上都与成人肿瘤有着显著差异[5,47]。目前大多数抗肿瘤靶向药物是针对成人适应证研究与开发的,并不能直接用于儿童肿瘤,仅有少数药物被FDA或欧洲药品管理局(European Medicines Agency,EMA)批准专门用于儿童肿瘤。因此,针对儿童肿瘤的药物研究需要系统地考虑儿童肿瘤的独特分子特征(见图1)。
2.1 儿童肿瘤相较于成人肿瘤基因突变率更低
与成人肿瘤相比,儿童肿瘤往往具有较低的基因突变负荷。针对儿童、青少年和成人的泛癌种分析结果表明,儿童中的不同肿瘤类型具有不同的基因突变频率(0.02 ~ 0.49 mutations/Mb),总体是成人肿瘤的1/14(儿童0.13 mutations/Mb,成人1.8 mutations/Mb)[5]。少数儿童肿瘤呈现相对较高的肿瘤突变负荷,主要包括具有mutS同源物2(mutS homolog 2,MSH2)和MSH6突变的H3K27野生型HGG以及骨肉瘤,尽管如此,上述基因突变仍低于同类型成人肿瘤中检测到的频率。
近年来一些研究猜想,儿童肿瘤的这种低突变负荷模式可能与儿童肿瘤的细胞起源或发病机制有关。成人肿瘤通常是环境致癌物质导致细胞突变累积造成的,而大多数儿童肿瘤起源于前体细胞或是发育过程中的失调细胞,因此突变累积相对较少[48]。实际上,大多数儿童肿瘤体细胞突变频率随着年龄增长而增加,且复发性肿瘤比原发性肿瘤呈现更高的突变负荷[5,47],这也提供了有力的证据证实这一猜想。
2.2 儿童肿瘤与成人肿瘤的基因突变谱显著不同
对儿童肿瘤和成人肿瘤中的体细胞变异分析发现,即使是在同一类型肿瘤中,两者的突变谱也明显不同。全基因组和全外显子组测序鉴定出142个儿童肿瘤中潜在的驱动基因,其中78个基因在成人肿瘤中从未被发现。而儿童肿瘤中的显著突变基因仅30%与成人肿瘤发生重合,并且组成类型和突变频率与成人肿瘤也明显不同[5]。儿童肿瘤中与表观遗传修饰相关的基因改变最为常见,其次是转录调节因子和MAPK相关的基因,在成人中则以转录调节因子为主。与PI3K相关的驱动基因改变在成人肿瘤中经常发生,而在儿童中仅占3%[5]。
值得注意的是,染色体结构变异在多种类型的儿童肿瘤中十分常见,尤其是导致融合基因产生的染色体易位,被认为是驱动多种儿童肿瘤发生发展的重要诱因[5,47]。比如一些融合基因在特定儿童肉瘤中发生频率较高,并被认为是患者不良预后和复发的高风险因素,其中典型的例子是尤文肉瘤中的EWS相关融合,以及横纹肌肉瘤中的PAX-FOXO1融合[39,49]。
2.3 儿童肿瘤存在单基因驱动性
与成人肿瘤中同时存在多个致癌突变不同,儿童肿瘤常由单基因改变事件驱动发生。泛癌种分析结果发现,大多数显著突变基因在不同类型的儿童肿瘤中是互斥的,不会同时存在,约57%儿童肿瘤仅存在1个显著突变基因[5]。相比之下,几乎大部分成人肿瘤中被发现至少存在1个显著突变基因。此外,儿童肿瘤中常伴有独特的基因融合特征,该特征在成人肿瘤中较少被发现。独特的单基因驱动模式可能为儿童肿瘤的治疗提供新的思路和策略,靶向这些突变或融合可能可以更加高效且低毒地干预儿童肿瘤的进展,因此有望成为非常有前景的治疗靶点[5]。
2.4 儿童肿瘤常起源于干细胞/祖细胞
儿童肿瘤与成人肿瘤之间另一关键的差异就是细胞来源不同。成人肿瘤大多来源于上皮细胞,通常由损伤和诱变引起突变累积导致。相比之下,儿童肿瘤多数来源于中胚层或神经外胚层,通常是由发育过程中未成熟或发育中的细胞发育受阻产生[48]。已有的一些证据证明了儿童肿瘤发育阻滞这种独特的生物学特性:许多肿瘤类型是儿童特有的,很少或从不在成人中出现,表明成熟组织中不存在这类儿童肿瘤的起源细胞,比如神经母细胞瘤、肝母细胞瘤等。即使一些肿瘤在成人和儿童中都能发生,但它往往在儿童中表现出独特的分子特征,比如儿童HGG主要是由体细胞组蛋白H3.3或H3.1突变驱动,而这种突变在成人中很少见[45]。另一关键证据是许多儿童肿瘤的发病率与年龄密切相关:神经母细胞瘤和肾母细胞瘤在0 ~ 4岁儿童中发生率较高,而在15 ~ 29岁的青少年中较少见;相反,骨肉瘤和尤文肉瘤等骨肿瘤在青少年中发病率达到高峰。以上这些证据提示儿童肿瘤与成人肿瘤的细胞来源不同,儿童肿瘤更多的来源于干细胞或祖细胞[2]。
综上所述,儿童肿瘤有着独特的分子特征,一方面儿童肿瘤常由单基因驱动,且整体基因突变率相较于成人肿瘤更低,这使得儿童肿瘤的分子靶向药物研究可以更有针对性,且更容易使儿童肿瘤患者获益。另一方面,儿童肿瘤与成人肿瘤的基因突变谱有着巨大差异,这使得原本应用于成人肿瘤的多数分子靶向疗法可能不完全适用于儿童肿瘤,因此,针对儿童肿瘤还亟需根据其独特的分子特征开发专门的分子靶向药物(见图1)。
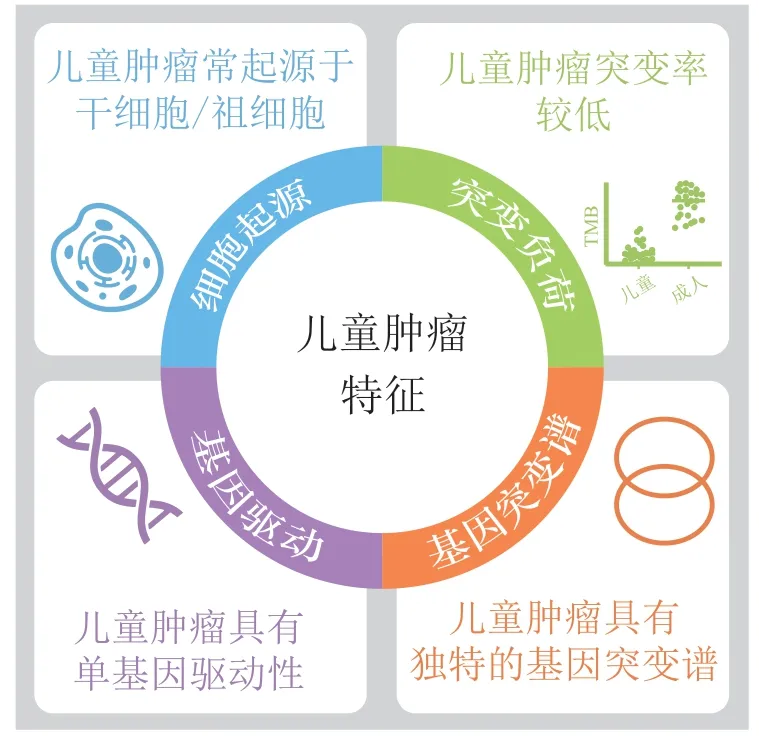
图1 儿童肿瘤的分子特征Figure 1 Molecular characteristics of pediatric tumors
3 儿童肿瘤分子靶向药物的研究进展与趋势
3.1 靶向致癌融合蛋白
致癌融合基因的产生是许多儿童肿瘤的重要特征之一,也是近年来研究关注的焦点。融合基因根据是否含有激酶结构域,可分为激酶型融合基因和非激酶型融合基因。激酶型融合基因通常会导致激酶的过度表达和持续激活,从而驱动肿瘤发生,因此多项研究集中在靶向融合蛋白的激酶结构域。最具代表性的例子是ABL激酶抑制剂在治疗具有BCR-ABL融合的ALL时取得巨大成功,这也是基于分子特征的精准医疗的重大发展转折点[50]。类似的治疗策略在儿童实体肿瘤中也取得了令人兴奋的结果。拉罗替尼(larotrectinib)是一种靶向NTRK的激酶抑制剂,已在Ⅰ/Ⅱ期临床试验中被证明可以有效治疗NTRK融合的儿童实体瘤,成为首个被用于治疗NTRK融合的儿童实体瘤的激酶抑制剂[51]。
激酶抑制剂在激酶型融合基因中取得的重大进展,为靶向融合基因的治疗策略提供了有利的证据,也为一些特定的儿童肿瘤类型提供了新的策略。然而相反的是,非激酶型融合基因的研究则面临着巨大挑战。非激酶型融合通常导致转录因子嵌合而使基因表达程序被异常驱动,且由于缺少可靶向的酶活口袋,小分子抑制剂难以直接作用于非激酶型融合蛋白。一种潜在的干预策略是降低融合蛋白的稳定性。早幼粒细胞白血病-维甲酸受体α融合蛋白(promyelocytic leukemia-retinoic acid receptor-α,
PML-RARα)被认为是导致急性早幼粒细胞白血病(acute promyelocytic leukemia,APL)发生的重要因素,研究发现三氧化二砷和全反式维甲酸能够结合PML-RARα融合蛋白,在临床试验中可以有效地改善APL患者的预后,已被纳入APL的标准治疗方案[52]。笔者课题组也首次鉴定APL的关键致癌驱动蛋白PML-RARα可发生蛋白质翻译后修饰Neddylation,阐明Neddylation修饰通过抑制相分离,从而干扰PML核体形成的全新APL致癌机制,并提出改变相分离重构核体的分化治疗新策略,发现MLN4924和G5等多个可克服APL耐药的先导小分子[53-54]。
此外,还有一些研究在尝试干预融合蛋白功能和下游途径。比如在尤文肉瘤中,BRG/BRM相关因子(BRG-/BRM-associated factor,BAF)复合物中的富含AT的相互作用结构域蛋白1A(AT-rich interactive domain 1A,ARID1A)核心亚基与EWSR1-FLI1融合蛋白相互作用,能够提高后者的蛋白稳定性从而导致其致癌能力增强。TK216抑制剂通过阻断EWSR1-FLI1与ARID1A相互作用来破坏融合蛋白功能,被认为有望成为复发性或难治性尤文肉瘤的潜在小分子,目前正在进行临床试验评估[55]。
3.2 靶向基因突变或扩增
RTK,RAS-MAPK和PI3K-AKT-mTOR等信号通路中的关键基因的突变或扩增,被发现与肿瘤的发生发展密切相关,可能成为潜在的干预靶点[5]。
儿童肿瘤中具有多种RTK通路的基因突变或扩增,包括ALK,成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR),KIT原癌基因(KIT proto-oncogene,KIT)和间质-上皮细胞转化因子(mesenchymal-epithelial transition factor,MET)的突变,以及血小板源性生长因子受体(platelet-derived growth factors receptor,PDGFR)和血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)的扩增等。ALK突变或扩增在NB中经常发生,被证明是导致NB恶性进展的重要因素。临床研究表明,具有ALK突变的NB患者对ALK抑制剂克唑替尼更加敏感。此外,ALK抑制剂在具有ALK融合蛋白的儿童肿瘤如ALCL中也展现出较好的活性。酪氨酸激酶抑制剂培唑帕尼(pazopanib)和瑞戈非尼(pegorafenib)正在进行临床试验评估,用于治疗儿童肿瘤[56-57]。
RAS-MAPK通路的异常激活也在多种儿童肿瘤类型中被检测到,其中关键的致癌驱动因素包括BRAFV600E突变、BRAF融合蛋白和NF1突变等[5]。临床研究表明,BRAF抑制剂达拉非尼(dabrafenib)和MEK抑制剂,在治疗具有BRAFV600突变的复发或难治性LGG儿童患者中,显示较好的缓解率[58]。PI3K-AKT- mTOR信号通路的激活在RMS的恶性演进中发挥重要作用,临床前研究已证明mTOR抑制剂能够显著抑制RMS细胞生长。目前,临床研究正在评估mTOR抑制剂坦罗莫司(temsirolimus)对中风险RMS的治疗作用[59]。
3.3 靶向表观遗传修饰
表观遗传的改变也是儿童肿瘤的主要特征之一,靶向表观遗传的改变是儿童肿瘤的潜在治疗策略之一。
组蛋白H3突变以及几种参与染色质重塑的蛋白质复合物改变,被发现与儿童肿瘤的恶性进展相关。SMARCB1(SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1)基因编码SWI/SNF(switch/sucrose nonfermentable)复合物中的核心亚基,该亚基参与核小体染色质重塑。基因组测序分析结果显示,在上皮样肉瘤和恶性RMS中存在SMARCB1失活突变,导致zeste基因增强子同源物2(enhancer of zeste homolog 2,EZH2)异常高表达[60]。临床研究表明EZH2抑制剂泰泽司他(tazemetostat)能够有效改善SMARCB1突变的上皮样肉瘤患者的预后[61]。此外,测序研究表明,T-ALL,B-ALL和AML中与表观遗传修饰相关的基因经常发生改变,包括组蛋白赖氨酸三甲基转移酶SETD2(set domain containing 2)、赖氨酸去甲基化酶6A(lysine demethylase 6A,KDM6A)、组蛋白去乙酰化酶2(histone deacetylase 2,HDAC2)和赖氨酸甲基转移酶2A(lysine methyltransferase 2A,KMT2A)重排[5]。KMT2A属于组蛋白修饰酶家族,其重排往往导致KMT2A丢失甲基转移酶活性。大多数KMT2A的融合伴侣参与调节转录延伸,或组成组蛋白甲基转移酶端粒沉默1-样干扰物(disruptor of telomeric silencing-1-like,DOT1L)复合物,研究表明KMT2A重排是导致白血病发生的重要因素之一[62]。目前,靶向具有KMT2A重排的治疗策略正在研究之中。Menin通过识别KMT2A的N端结构域与融合蛋白相互作用,从而影响造血干细胞的增殖能力与分化能力[62]。SNDX-5613抑制剂能够阻断Menin和KMT2A的相互作用,从而抑制白血病干细胞的增殖,并且诱导重新开始分化[63]。DOT1L也是一种潜在治疗靶点,匹诺司他(pinometostat)是DOT1L辅因子S-腺苷甲硫氨酸(S-adenosylmethionine,SAM)的竞争性抑制剂,能够有效干预DOT1L的催化功能[64]。以上结果表明靶向表观遗传修饰的异常改变是儿童肿瘤的潜在治疗策略。
3.4 靶向分化障碍
很多儿童肿瘤存在分化障碍的特征,分化疗法是使用分化诱导药物使肿瘤细胞走向终末端分化变成正常成熟细胞的肿瘤治疗方法,具有较好抗肿瘤效果[65]。
分化疗法用于肿瘤治疗的典型成功案例是维甲酸(retinoic acid,RA)类药物和三氧化二砷用于治疗APL,目前该方案是APL的临床标准治疗方案。RA是分化的主要调控因素,可以激活RARα等转录因子从而促进细胞分化的发生。在APL中,RARα与PML形成融合蛋白PML-RARα,一方面增强与转录共抑制子的结合从而导致RARα依赖的分化基因的转录抑制,另一方面抑制PML核体的形成,可促进APL恶性肿瘤细胞的自我更新,从而导致APL的发生。RA类药物发挥功能的主要机制是与PML-RARα融合蛋白的RARα部分结合,解除分化相关基因的转录抑制,诱导分化的发生。三氧化二砷则主要与PML-RARα的PML部分结合,虽然不能调控RARα依赖的分化转录因子,但仍能诱导分化表型[65-66]。
分化疗法目前在儿童血液系统肿瘤中取得良好应用,除RA类药物和三氧化二砷外,环磷酸腺苷(cyclic adenosine monophosphate,cAMP),维生素D3和IDH抑制剂等被发现可以用于诱导AML分化,部分已进入临床试验[65]。此外,笔者课题组在国际上率先提出“降解细胞周期蛋白依赖性激酶2(cyclin-dependent kinase 2,CDK2)诱导细胞分化”的AML治疗新策略,并发现CDK2调控AML细胞分化的机制完全独立于经典靶点RARα[67]。基于此,课题组与清华大学饶燏教授合作设计合成了首个CDK2选择性蛋白降解靶向嵌合体(proteolysis targeting chimeras,ROTAC)降解剂,为儿童AML患者的分化治疗提供了新靶点及崭新分子实体[68]。
在儿童实体瘤中,大量研究表明RA类药物可以诱导NB和骨肉瘤细胞分化,在这2个癌种中发挥抗肿瘤作用[69-70]。笔者课题组还发现在骨肉瘤中E3泛素连接酶鼠双微基因2(murine double minute 2,MDM2)可降解RARα限制RA的分化效果,因此,MDM2抑制剂联用RA可明显促进骨肉瘤的分化[71]。此外,曲贝替定(trabectedin)、地西他滨(decitabine)等药物也被发现可以诱导骨肉瘤细胞分化,目前已进入临床试验[69]。笔者课题组发现RA类药物可通过抑制巨噬细胞M2型极化,发挥抗骨肉瘤转移的新作用,为分化药物应用于骨肉瘤儿童患者的临床治疗提供崭新的视角[72-73]。
综上所述,目前围绕致癌融合蛋白、基因突变或扩增、表观遗传修饰以及细胞分化等的儿童分子靶向药物研究取得了巨大进展。这些分子靶向药物从儿童肿瘤特有的分子特征出发,在临床前体内外实验中展现出显著的抑制肿瘤效果,且部分已进入临床试验(见表2)。然而目前儿童肿瘤分子靶向药物的研究在癌种上仍存在一定局限性,主要以血液肿瘤、肉瘤及神经系统肿瘤为主。针对其他儿童肿瘤癌种的融合、突变及扩增等分子特征还有待进一步研究和完善,以便于针对这些癌种开发分子靶向药物。

表2 儿童肿瘤中潜在可靶向的分子靶点及临床研究进展[46]Table 2 Potential targeted therapies in pediatric tumors and their clinical research progress
4 总结与展望
综上所述,儿童肿瘤是威胁儿童生命健康的主要疾病之一,目前分子靶向药物展现出较好的应用前景,但儿童肿瘤的临床治疗仍亟需药物新靶点和新策略。儿童肿瘤不能与成人肿瘤一概而论,儿童肿瘤具有独特的基因组“景观”,这为分子靶向药物的研发提供了思路也带来了挑战。与成人肿瘤相比,儿童肿瘤往往呈现较低的肿瘤突变负荷,在这种相对简单的基因背景下,特定突变或融合事件发挥了关键致癌驱动力的作用,为针对儿童肿瘤的药物开发提供了新的机遇。大规模的下一代测序技术极大程度提高了研究者对儿童肿瘤发病机制的理解,也为临床上诊断监测、制定个体化的治疗方案提供了新思路。目前儿童肿瘤中许多重要的分子改变仍缺少有效的靶向干预手段,因此未来还需要针对这些基因改变,开展系统的机制研究和药物研发,以期为儿童肿瘤患者提供更好的靶向治疗策略。

续表2
