自噬在抗肿瘤药物毒性中的作用与机制研究进展
2022-02-09陈剑曾艳高子正胡誉怀颜皓徐志飞罗沛华杨波何俏军
陈剑 ,曾艳 ,高子正 ,胡誉怀 ,颜皓 ,徐志飞 ,罗沛华 ,杨波 ,何俏军
(1. 浙江大学药学院,浙江 杭州 310058;2. 浙江大学智能创新药物研究院 浙江省抗肿瘤药物临床前研究重点实验室,浙江 杭州 310018)
早在1962年,Ashford 和 Porter等学者在人肝细胞中便观察到了自噬现象,并在1963年由Christian de Duve提出自噬(autophagy)的概念。当细胞处于营养缺乏或应激条件下时,细胞会将胞质中的蛋白或受损细胞器通过囊泡包裹并运送到溶酶体降解,从而维持能量供应和清除有害物质[1]。自噬的发生机制十分复杂,目前已有40余个自噬相关基因(autophagy-related genes,ATG)被报道参与自噬体的形成过程,此外自噬还受到多种上游蛋白的协同调控[2]。自噬在生物体的生理病理进程中发挥了重要的作用,如参与生物体生长发育、代谢调节以及衰老等过程,同时也与肿瘤的形成、神经退行性疾病、心血管系统疾病、克罗恩病和肌病等疾病密切相关[3]。此外,细胞自噬在药物诱导的毒性过程中也发挥重要的作用,机体内自噬的功能或活性异常可诱导脏器毒性。本文对自噬调控机制、生理病理作用及在抗肿瘤药物毒性中的作用与机制进行了综述。
1 自噬的基本概念
细胞自噬是细胞的一种自我降解途径,可以将大量的胞质蛋白或细胞器输送到哺乳动物细胞的溶酶体或植物和酵母细胞的液泡中进行降解[4]。这种分解代谢过程在真核细胞的进化过程中高度保守,对于防止细胞损伤、在能量或营养匮乏的情况下维持细胞的存活具有重要意义[5]。在对自噬过程的研究中,目前已发现3种将物质递送到溶酶体中进行降解的转运模式,由此可将自噬归纳为3种类型:巨自噬(macroautophagy)、微自噬(microautophagy)和分子伴侣介导的自噬(chaperone-mediated autophagy)。巨自噬是最常见的自噬形式,在此过程中,细胞会产生双层膜结构,包裹胞内的异常蛋白或受损细胞器形成自噬体,自噬体最终会与液泡或溶酶体融合并降解其中的“货物”;在微自噬过程中,液泡膜或溶酶体膜会直接内陷并吞噬边界膜上的细胞质“货物”;由分子伴侣介导的自噬其底物具有特定序列,由热休克蛋白70(heat shock protein 70,HSP70)和其他分子伴侣蛋白介导,与溶酶体上的受体LAMP-2A结合从而进入溶酶体降解自噬底物[6-7]。巨自噬是调控环境变化和生理信号的主要自噬形式,无特殊说明,本文中提到的自噬均指巨自噬。
2 自噬的过程和分子机制
2.1 自噬的过程
细胞自噬的过程可分为以下几步:1)在自噬的启动阶段,酵母细胞中会形成自噬体前体结构(preautophagosomal structure,PAS)并招募其他Atg蛋白和囊泡等膜成分定位,而在哺乳动物细胞内质网上会形成欧米茄体(omegasome)并和分离膜形成杯状结构。2)在多种Atg(酵母细胞)/ATG(哺乳动物细胞)蛋白的协助下,自噬体的双层膜延伸并包裹各种胞内成分,最终封闭形成自噬体。3)自噬体的外膜与哺乳动物细胞中溶酶体或酵母中的液泡膜融合,形成自噬溶酶体;内膜和其中的包含物被溶酶体内的蛋白酶、脂肪酶等分解为小分子,降解产物会返回细胞质被重新用于构建新的分子,这在细胞的新陈代谢中发挥重要作用[2,5,8](见图1[9])。
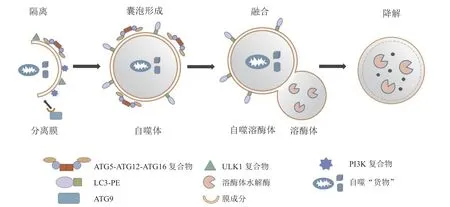
图1 自噬的发生过程Figure 1 The process of autophagy
2.2 自噬的分子机制
自噬过程从启动到终止受到多种蛋白质的协同精密调控,目前在酵母中已鉴定出了40余种ATG基因,其中大多数在哺乳动物基因组之间表现出显著的同源性[2]。在众多基因当中,约有18个核心基因编码的蛋白质在细胞自噬过程中起关键的调控作用[10],例如:1)Atg1/unc-51样激酶(unc-51-like kinase,ULK)复合体;2)含Atg9/ATG9A的囊泡;3)磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)复合体;4)类泛素化结合系统。
2.2.1 Atg1/ULK复合体Atg1是酵母细胞中的一种自噬特异性的激酶,Atg1,Atg13,Atg17,Atg29和Atg31共同组成了Atg1 核心复合体,在自噬体的形成过程中起关键作用。在营养丰富的条件下,雷帕霉素复合物1(target of rapamycin complex 1,TORC1)与Atg13紧密结合并使其磷酸化,阻断其与Atg1和Atg17-Atg31-Atg29复合物的相互作用[11-13]。在营养剥夺或用雷帕霉素处理抑制TORC1后,Atg13会迅速脱磷酸化并与Atg1结合,这使得Atg1激酶活性上调,发生自身磷酸化并形成二聚体,促进分离膜的形成[14-15]。哺乳动物细胞中与Atg1同源的蛋白是ULK1/2,ULK1/2与ATG13、相对分子质量为200 000的黏着斑激酶家族相互作用蛋白(focal adhesion kinase family interacting protein of 200 000,FIR200)和ATG101构成ULK1/2复合体[16]。正常情况下,TORC1能够与ULK1/2复合物结合使其磷酸化来抑制其激酶活性,而在能量匮乏的情况下,TORC1活性降低,ULK1/2被激活并触发ATG13,ULK和FIP200的磷酸化[17]。激活的Atg1/ULK复合体可直接磷酸化酵母中的Atg4和Atg9,以及哺乳动物细胞中ATG4B,ATG9,ATG14L和PI3K复合物的亚基BECLIN1等多种蛋白质,从而驱动自噬的发生。
2.2.2 含Atg9/ATG9A的囊泡Atg9/ATG9A蛋白是自噬相关蛋白中唯一的跨膜蛋白,研究表明其在引导供体细胞器膜以形成自噬体过程中起关键作用。在酵母细胞中,Atg9位于细胞质的移动囊泡上,这些囊泡定位于特定的区域,可以看做是Atg9在胞质中的贮存库,自噬发生时,Atg9会在酵母PAS和胞质的储存库之间来回转运,为自噬体的形成提供脂质和蛋白质,为分离膜扩张提供原料,这个过程需要Atg23和Atg27等蛋白的协助[18-19]。在哺乳动物细胞中,ATG9A(Atg9的同源物)的细胞内定位更为复杂,ATG9A囊泡在反式高尔基体网状结构(trans Golgi network,TGN)中产生,TGN与ATG9A隔室之间的循环中包含了丰富的小管和囊泡簇。在能量匮乏的情况下,ATG9A可依赖ULK1和PI3K的活性从反式高尔基体转移到晚期内体和自噬泡上。ATG9A也定位于质膜,从中产生ATG9A囊泡并运输到循环内体,ATG9A隔室和循环内体都被认为是自噬体膜的来源[20-22]。
2.2.3 磷脂酰肌醇3-激酶复合体PI3K是一类重要的信号调控分子。根据结构特征和基质特异性,PI3K可以分为3类,其中第Ⅲ类的PI3K复合物(class III PI3K complex,PI3KC3)可磷酸化膜上的磷脂酰肌醇(phosphatidylinositol,PI)来调节细胞的生长、分化和物质运输[23],同时也是自噬体形成过程中的主要蛋白复合物。酵母细胞中的PI3K复合物由膜泡分拣蛋白15(vacuolar protein sorting 15,Vps15),Atg6,Vps34,Atg14和Atg38组成[24], 自噬发生时,Atg6介导与Atg14的相互作用,将PI3KC3定位于PAS上,复合物中的Vps34能够激活PI3K,催化PI磷酸化形成磷脂酰肌醇3-磷酸(phosphatidylinositol 3-phosphate,PI3P),自噬膜上的PI3P可结合膜结合蛋白Atg18并将其募集到双层膜上,故其对于自噬体的伸长和完成至关重要[25]。在哺乳动物细胞中,PI3K复合物参与自噬体的形成及晚期的成熟和转运。Atg6的同源蛋白BECLIN1可与VPS34和VPS15结合形成核心复合物[26],自噬发生时,BECLIN1-VPS34-VPS15可与ATG14L结合形成ATG14L-BECLIN1-VPS34-VPS15复合物参与自噬体的形成,还可与紫外线抵抗相关基因(UV radiation resistance-associated gene,UVRAG)编码的蛋白结合形成UVRAG-BECLIN1-VPS34-VPS15复合物,在自噬体的成熟和转运中产生重要影响[27-28]。
2.2.4 类泛素化结合系统细胞自噬过程中存在2个与泛素修饰系统相似的蛋白质结合系统,这2种结合体系在进化上都是保守的,在自噬体的延伸过程中发挥重要的作用。其一是Atg12连接体系,在自噬进程中泛素活化酶(ubiquitin-activating enzyme,E1)样酶Atg7以ATP依赖的方式与Atg12羧基末端的甘氨酸残基结合并将它激活,接着将Atg12转移至泛素结合酶(ubquitin-conjugation enzyme,E2)样酶Atg10,Atg10有类似E2的作用,介导Atg12与Atg5的赖氨酸残基结合,再与Atg16结合形成Atg12-Atg5-Atg16复合物[29]。Atg16含有卷曲螺旋结构域,可以自身结合,最终构成2 : 2 : 2的复合体,此多聚复合物为自噬过程所必需[30]。其二是Atg8连接体系,首先Atg8的C端被蛋白酶Atg4剪切,末端变为甘氨酸,之后E1样酶Atg7将Atg8传递到E2样酶Atg3,在Atg12-Atg5-Atg16复合物的协助定位下,最终与双层膜上的磷脂酰乙醇胺(phosphatidyl ethanolamine,PE)结合,帮助自噬体双层膜的扩展[31-32]。在哺乳动物细胞中,LC3是Atg8的同源蛋白,被hATG4B剪切变成LC3-Ⅰ,再同样经过ATG7和ATG3的协助与膜上的PE结合,成为膜结合形式LC3-Ⅱ[33]。LC3-Ⅱ与自噬体膜紧密结合,当与溶酶体融合后则会被降解,因此常将LC3-Ⅱ作为自噬体的表面标记物[34]。
2.3 自噬的调控机制
2.3.1 依赖哺乳动物雷帕霉素靶蛋白的调节通路哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)属于丝氨酸/苏氨酸激酶,参与细胞发育、核糖体生成和代谢调控等生物学过程。mTOR以2种形式存在:mTORC1和mTORC2,前者主要在细胞生长、凋亡和自噬中发挥作用,后者调控细胞骨架蛋白的构建和存活[35-36]。mTORC1通过磷酸化Atg1/ULK复合体使其失活,对细胞自噬起负调控作用,可被雷帕霉素阻断;mTORC2对雷帕霉素不敏感,不直接参与自噬调节。一般认为,mTOR是调节细胞生长、增殖和自噬等上游信号转导通路的聚集点,如腺苷酸活化蛋白激酶(AMPactived protein kinase,AMPK)、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)和PI3KC1等多种信号[37]。
1)腺苷酸活化蛋白激酶信号通路
AMPK是一种丝氨酸/苏氨酸蛋白激酶,能够感知AMP/ATP的变化,在细胞中充当能量传感器。在各种生理和病理条件下,AMPK可被上游激酶磷酸化,并与AMP或ADP结合导致其活化,从而调节各种代谢过程[35]。AMPK通过磷酸化mTORC1,ULK1和PIK3C3/VPS34复 合 物 等自噬相关蛋白,或通过调节转录因子[如叉头框蛋白O3(Forkhead box O3,FOXO3),转录因子EB(transcription factor EB,TFEB)和溴结构域蛋白4(bromodomain-containing protein 4,BRD4)]调控下游ATG的表达,直接促进自噬发生。此外,由于AMPK可诱导受损线粒体的片段化,并促进自噬体向受损线粒体的易位,其还可上调线粒体的自噬降解水平[38-39]。
2)丝裂原活化蛋白激酶信号通路
与AMPK类似,MAPK作为另一类重要的丝氨酸/苏氨酸蛋白激酶,参与细胞的生长、分化、对环境的应激适应和炎症反应等多种重要的生理/病理过程。MAPK信号通路有4条主要的分支路线,分别为:细胞外调节蛋白激酶(extracellular regulated protein kinase,ERK)、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)、P38 MAPK和ERK5。MAPK/ERK信号通路可被受体酪氨酸激酶激活,活化的ERK二聚体可直接激活mTOR或通过影响结节性硬化复合物(tuberous sclerosis complex,TSC)1/2的形成调控mTOR,从而影响自噬[40]。另有研究表明,JNK通路的激活可诱导癌细胞的自噬和凋亡,P38 MAPK信号通路可介导内质网应激途径诱导人牙龈细胞自噬[41]。
3)PI3K/AKT/mTOR信号通路
PI3K/蛋白激酶B(protein kinase B,PKB,即AKT)/mTOR信号通路是调控细胞生命活动的经典通路之一,参与调控细胞分裂、分化、凋亡、能量代谢和肿瘤发生等过程。PI3K/AKT是mTOR上游的主要调节剂,当Ⅰ类PI3K被激活后,它能作用于磷脂酰肌醇-4,5-二磷酸[phosphatidylinositol (4,5) bisphosphate,PIP2]并 使 其 转 变 为PIP3,PIP3募集AKT和3-磷酸肌醇依赖性蛋白激酶-1(3-phosphoinositide-dependent protein kinase 1,PDK1)到细胞内膜附近,同时AKT蛋白的Ser308位点被磷酸化而激活。活化的AKT可以磷酸化TSC1/2并使RAS蛋白脑组织同源类似物(RAS homolog enriched in brain,RHEB)与三磷酸尿苷水解酶(GTP酶)结合最终促进mTOR的累积和活化,从而抑制自噬的发生。此外,AKT也可直接通过磷酸化mTOR分子来抑制自噬[42]。
4)P53信号通路
P53是一种肿瘤抑制因子,在细胞中可被广泛的应激条件激活,例如DNA损伤、缺氧或异常癌基因表达,以促进细胞周期检查点、DNA修复、细胞衰老和凋亡。有关P53的研究多集中在其与凋亡的关系上,但近年的文章也发现了它与自噬间的关系。在营养缺乏或毒性的刺激下,P53可以通过激活AMPK或通过DNA损伤调节自噬调控因子(DNA damage regulated autophagy modulator,DRAM)的反式激活抑制mTOR并刺激自噬[43-45]。另一个促进自噬的P53靶基因是ISG20L1,ISG20L1的过表达能促进自噬和细胞死亡[46]。
2.3.2 不依赖mTOR的调节通路BECLIN1是组成PI3KC3复合物的蛋白之一,在自噬形成的早期阶段起到重要的作用。然而,在自噬过程中BECLIN1除了能与PI3KC3结合外,还能与UVRAG,Bax相互作用因子1(Bax interacting factor 1,BIF-1)和BECLIN1调节自噬的激活分子(activating molecule in BECLIN1 regulated autophagy,AMBRA)等蛋白结合,并正向调节PI3KC3复合物的活性来激活自噬。此外,BECLIN1还能与抗凋亡蛋白BCL-2[B细胞淋巴瘤/白血病2基因(B-cell lymphoma 2,BCL-2)编码产物]以及BCL-XL结合,所形成的复合物具有抑制自噬信号的功能[47-48]。
3 自噬在生理病理中的作用
3.1 自噬的生理作用
细胞自噬广泛存在于人体正常细胞的生理过程中,总体来说,自噬介导了细胞中的几种生物功能,例如细胞质物质的清除、降解产物的产生以及物质从细胞质到溶酶体的转运[49-50]。自噬最基本的作用是适应机体的代谢需求,在饥饿或能量需求增加的情况下,细胞通过自噬溶酶体降解产物的再利用维持正常能量供应和蛋白质的合成。实验表明,新生的小鼠在中断胎盘营养供应后可通过上调细胞自噬水平来维持生存[51]。同时自噬能起到维持细胞稳态的作用,在一些长寿细胞群中,细胞不能够通过增殖来更新换代,细胞内产生的一些有害物质如异常蛋白质聚集体或损伤的细胞器可通过自噬途径清除,以维持细胞的正常状态。此外,在病原体感染过程中,自噬通过降解细胞内细菌和病毒来帮助免疫反应。还有研究表明自噬有助于延长哺乳动物的寿命和“健康跨度”[52]。例如在正常或适度应激的心肌细胞中,自噬可以降解失活的蛋白质为氨基酸,作为心肌细胞生存和发育的物质来源之一;在缺氧的条件下,自噬可以增加ATP的合成,维持心肌细胞的能量代谢;另外,心肌细胞富含线粒体,在不良条件下线粒体容易发生损伤,释放促凋亡因子如细胞色素c诱导细胞凋亡,而自噬能够清除受损线粒体阻断凋亡,保护心肌细胞免受损伤[53]。
3.2 自噬的病理作用
在大多数情况下,自噬作为一种重要的细胞保护机制发生在较低的基础水平,能在不利条件下提高细胞抗压能力并维持存活,而自噬的功能破坏与许多疾病的发生发展存在相关性。动物实验的结果显示自噬关键编码基因敲减或缺失小鼠的自噬水平受到抑制,导致机体丧失自我调节的能力,无法应对外界生存环境中的变化。对人类来说,自噬功能障碍会导致许多疾病的发生发展,包括癌症、心血管系统疾病、克罗恩病和神经退行性疾病如阿尔茨海默病、亨廷顿病和帕金森病等。
3.2.1 癌症自噬在肿瘤中发挥的作用具有双向性,在肿瘤发展的早期阶段,自噬可以防止肿瘤发生并抑制癌症进展,然而在肿瘤发展的后期阶段或是在应激条件下,自噬可以作为一种保护机制促进肿瘤的发展[23]。BECLIN1是在细胞自噬中发挥重要作用的蛋白,在75%的人类肝癌、乳腺癌、卵巢癌和前列腺癌病例中都能观察到BECLIN1基因的缺失,Qu等[54]采用基因敲除的方法建立Beclin1+/-小鼠模型,发现Beclin1单等位基因缺失可以增加自发性恶性肿瘤的频率,并且加速了乙型肝炎病毒诱导的癌前病变的发展,说明Beclin1基因缺失导致的自噬水平下降可促进癌症的发展。在另一些癌症如胃癌、结直肠癌、宫颈癌患者组织中可以观察到BECLIN1自噬基因的上调,这是由于在应激条件下,自噬可以帮助肿瘤细胞去除受损的线粒体、抑制DNA损伤、维持基因组稳定、限制炎症,从而有助于肿瘤细胞的存活[23]。以上结果说明自噬与肿瘤的发生和发展密切相关,但在不同的肿瘤或肿瘤发展的不同阶段,自噬所起到的作用会存在差别。
3.2.2 心血管系统疾病心血管系统疾病包括缺血/再灌注损伤(ischemia-reperfusion injury,IRI)、心肌梗死、心肌肥厚和心力衰竭等,心肌细胞的死亡是诱发此类疾病的关键因素,而自噬调控在心血管系统疾病中发挥重要作用[55]。自噬过程的抑制是心功能障碍和重构心肌细胞死亡的关键原因,在多种心脏病中都伴有自噬活性的降低[56]。在斑马鱼心肌细胞中特异性敲除atg5,atg7和beclin1,会导致斑马鱼心脏缺陷,并降低斑马鱼生存率。心肌细胞特异性缺失atg5会引起心肌肥厚和功能障碍,心肌细胞特异性缺失atg7会引起心脏收缩功能障碍[57]。而自噬体-溶酶体融合所需的溶酶体相关膜蛋白LAMP-2的全身敲除会导致自噬空泡的积累,降低小鼠的心脏收缩能力和生存率,IRI可通过损害自噬体-溶酶体融合以及损害自噬小体清除引起心肌细胞死亡[58]。另一方面,非适应性的自噬水平可能会通过影响关键底物蛋白的降解导致心肌细胞死亡。苏尼替尼会诱导心肌细胞关键蛋白细胞通信网络因子2(cellular communication network factor 2,CCN2)的过度自噬降解,从而导致心肌细胞死亡[59-60],自噬抑制也可能通过促进有害蛋白的积累而导致心肌细胞死亡和心功能障碍。这说明自噬活性的精细调控对正常心功能和内稳态是必需的,靶向自噬活性异常可能是对抗自噬相关心血管系统疾病的可行策略。
3.2.3 克罗恩病克罗恩病是一种胃肠道的慢性非特异性炎症性肠病,其发病原因与多种因素有关,包括Paneth细胞功能障碍、内质网应激、炎症反应和自噬缺陷等。在克罗恩病与自噬相关的研究中,自噬基因ATG16L1被认为是克罗恩病的风险等位基因[61],ATG16L1基因N端区域(在酵母ATG16中保守)和C端WD重复结构域之间的T300A突变增加了克罗恩病的风险。Atg16L1基因缺失或Atg16L1T300A敲入小鼠具有各种异常表型,例如巨噬细胞中促炎细胞因子的释放增强,Paneth细胞的颗粒分泌减少,对沙门菌感染的易感性增加以及T细胞免疫失调,这可能都与克罗恩病的发生相关[62-64]。
3.2.4 神经退行性疾病神经退行性疾病是年龄依赖性遗传性或散发性疾病,表现为神经功能退行性丧失。线粒体功能障碍或由于突变和清除机制受损而导致的蛋白质聚集体积累是引发神经退行性疾病的主要原因。神经元是不可再生细胞,无法通过细胞分裂去除受损的细胞,而细胞自噬作为清除异常蛋白质的方式可以防止神经元的损伤。在小鼠模型中,特异性敲除神经元细胞的自噬基因会使泛素化蛋白包涵体的堆积增多,诱发神经退行性疾病[65-66]。同样,神经退行性疾病患者的自噬水平也发生失调,如帕金森病患者的多巴胺能神经元中α-突触核蛋白错误折叠和聚集,而这种蛋白的蓄积具有细胞毒性,可能通过阻断内质网-高尔基体水泡运输而阻碍自噬。在亨廷顿病中,亨廷顿蛋白基因中CAG三核苷酸的序列发生重复并扩增,使翻译出的蛋白质含有膨胀的聚谷氨酰胺束,最终错误折叠形成致病构象。在治疗手段上,增强神经退行性疾病患者的自噬水平已被证明可以减少异常蛋白质的积累并起到治疗作用[49,67]。
4 自噬在抗肿瘤药物毒性中的作用与分子机制
肿瘤的治疗一直是医药学领域的研究热点和难点,恶性肿瘤即癌症已成为导致人类死亡的主要原因之一,对人类的生命健康造成了很大的威胁。目前临床上治疗恶性肿瘤的方法以化疗为主,这些化疗药物包括烷化剂类药物、抗肿瘤抗生素、抗肿瘤激素类药物、金属铂类药物、抗代谢药物和抗肿瘤植物药等[68]。然而,抗肿瘤药物的连续、大量使用,在杀死肿瘤细胞的同时可能也会造成正常细胞的损伤而产生毒性,包括心脏毒性、肺毒性、肝脏毒性、胃肠道毒性、血液毒性和神经毒性等[69]。这些药物相关毒性限制了抗肿瘤治疗过程中的药物使用剂量和疗程,降低了患者的总体预后与生存质量。因此,通过研究抗肿瘤药物引起毒性的分子机制,找到合适的干预策略以降低毒性并提高疗效成为一个亟待解决的问题。目前许多有关抗肿瘤药物引起脏器毒性机制的研究表明,自噬在其中起到了重要作用,某些抗肿瘤药物会破坏机体正常的自噬水平,这可能引发保护性自噬或导致致死性自噬的发生。
4.1 抗肿瘤药物的毒性诱发机体产生保护性自噬
某些抗肿瘤药物在应用后其副作用会导致脏器的损伤,在这种条件下机体可能会激活自身的保护性自噬,这有助于不良因素的清除,维持细胞在恶劣条件下的存活(见图2)。
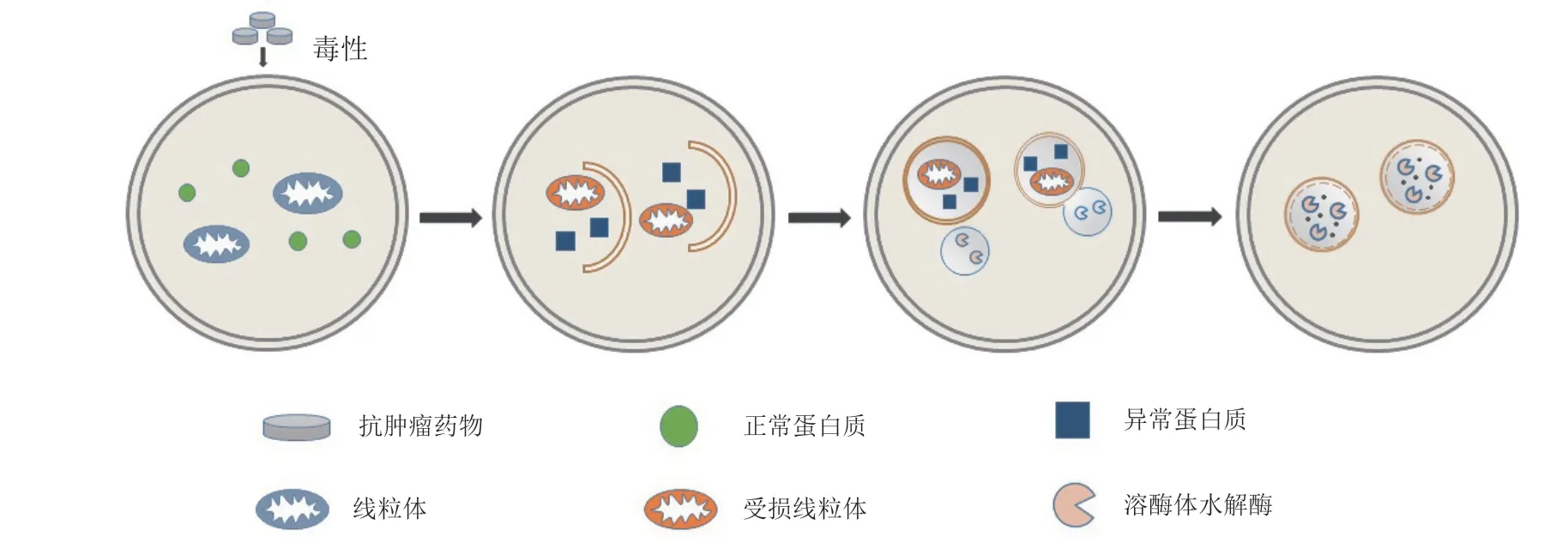
图2 保护性自噬的机制Figure 2 Mechanisms of protective autophagy
蛋白激酶抑制剂达沙替尼在临床上用于治疗慢性髓细胞性白血病(chronic myelogenous leukemia,CML)。然而该药具有较强的肝毒性,在CML患者中超过半数患者体内谷丙转氨酶和谷草转氨酶含量升高,此外还有少数伴有胆红素的升高[70]。Yang等[71]研究发现达沙替尼在导致大鼠肝细胞毒性的同时也升高了肝细胞中LC3-Ⅱ的水平,表明其诱导了自噬。若通过PI3KC3抑制剂3-MA或siRNA沉默Atg5来抑制细胞自噬,则会增加达沙替尼引起的肝细胞凋亡,由此说明达沙替尼自身诱导的自噬对肝细胞具有保护作用,若激活肝细胞的自噬水平则有利于改善药物的肝毒性。后续的实验表明达沙替尼通过活化P38-MAPK通路激活自噬,而P38可作为改善肝毒性的潜在靶点。盐酸异丙肾上腺素(isoproterenol,ISO)具有激活P38的作用,对肝细胞合用达沙替尼和ISO后结果显示细胞的自噬水平增强、细胞死亡数减少,证明ISO可通过激活P38诱导自噬来减少达沙替尼对肝脏的毒性作用。
顺铂是一种含铂的抗癌药物,临床上对卵巢癌、前列腺癌、睾丸癌、肺癌、鼻咽癌和食道癌等多种癌症具有疗效。然而,25% ~ 30%的顺铂治疗患者可能会出现肾毒性,如急性肾损伤,限制了其临床应用[72]。Zhu等[73]研究发现用顺铂处理48 h的HK2细胞中LC3-Ⅱ水平升高,而P62水平降低,在合用自噬激活剂雷帕霉素或拉帕替尼后观察到细胞活力增加,而合用自噬抑制剂羟氯喹后细胞活力进一步降低,表明自噬在顺铂诱导的细胞损伤中起保护作用。先前的研究表明海藻糖可改善多囊肾和Akt2敲除诱导的小鼠胰岛素抵抗模型中的肾功能[74], Zhu等[73]进一步研究发现海藻糖可通过增加自噬转录调节剂TFEB的活化来增强自噬,协同机体自身的保护性自噬减轻顺铂诱导的急性肾损伤。
4.2 抗肿瘤药物过度激活自噬导致脏器损伤
4.2.1 自噬进程通畅,导致关键蛋白降解,引发损伤有的抗肿瘤药物在发挥治疗作用的同时可能会诱导细胞自噬水平的升高,过度的自噬会导致关键蛋白被降解,影响细胞正常功能,或通过促进细胞凋亡,从而产生毒性(见图3)。

图3 抗肿瘤药物相关毒性的发生机制Figure 3 Mechanisms of anti-tumor drug-related toxicity
苏尼替尼是一种小分子多靶点的酪氨酸激酶抑制剂,对肾细胞癌、胃肠间质瘤等多种肿瘤具有治疗作用,但其具有显著的心脏毒性,会造成左心室功能障碍和心肌细胞的死亡。Xu等[59]研究发现经苏尼替尼处理后,心肌细胞中LC3-Ⅱ的表达呈时间和浓度依赖性增加,表明苏尼替尼升高了心肌细胞的自噬水平,而自噬的激活使得心脏中的一种细胞通信相关的关键蛋白CCN2通过自噬溶酶体途径降解。CCN2蛋白在心脏中高度表达,但其生理病理学作用复杂,目前尚未完全阐明,但感染带有靶向CCN2的siRNA的腺相关病毒9(adeno-associated virus 9,AAV9)以敲低小鼠心脏CCN2水平可导致心肌细胞凋亡和心功能障碍,说明了CCN2与心脏损伤的相关性。此外,在小鼠心肌细胞中特异性杂合敲除Atg7能够逆转苏尼替尼导致的心脏损伤。以上结果说明苏尼替尼能够激活心肌细胞的自噬,使关键蛋白CCN2通过自噬途径降解,从而引发了心脏损伤。
吉非替尼是非小细胞肺癌临床使用最为广泛的药物,然而在接受吉非替尼治疗的患者中,高达50% ~ 70%的患者出现了肝损伤,影响了吉非替尼治疗的总体疗效。Luo等[75]通过实验发现吉非替尼的应用增加了肝细胞中凋亡标记物多聚(ADP-核糖)聚合酶(裂解的)[ cleaved poly(ADP-ribose)polymerase,c-PARP]和自噬标记物LC3-Ⅱ的表达水平,GFP-mCherry-LC3转染细胞检测自噬流发现绿色荧光消失,红色荧光保持稳定,说明吉非替尼激活了肝细胞的自噬和凋亡;对线粒体数量的检测显示吉非替尼对线粒体并没有影响,因此推测某些关键蛋白质的降解可能是肝损伤的原因;通过蛋白质组学的方法比较对照组、吉非替尼组和吉非替尼合用氯喹组,筛选出3种差异表达的蛋白质,其中细胞色素c氧化酶6A1(cytochrome c oxidase subunit 6A1,COX6A1)的表达量受吉非替尼时效和量效影响最大;为了验证COX6A1通过自噬降解,作者用免疫荧光的方法发现在吉非替尼加氯喹的作用下COX6A1与mCherry-GFP-LC3B发生共定位,表明COX6A1存在于自噬体中;用siRNA构建COX6A1缺陷的肝细胞发现这促进了肝细胞的凋亡。这些证据表明吉非替尼诱导肝细胞自噬过度激活,降解了关键蛋白COX6A1,这种蛋白的缺失导致肝细胞凋亡的增加,由此产生了肝脏毒性。
4.2.2 自噬进程阻断,导致自噬体累积、蛋白质异常累积,引发损伤若抗肿瘤药物影响自噬的进程,如抑制自噬体溶酶体的融合,或是降低溶酶体的降解功能,则会导致自噬不能正常清除细胞内的异常蛋白质或受损细胞器,由此会对细胞产生毒性。
伊马替尼是首个被批准的酪氨酸激酶抑制剂,在临床可用于治疗CML、急性淋巴细胞白血病、胃肠道间质肿瘤和肥大细胞增多症等,然而伊马替尼的使用可能会引起不良反应,如水肿、肌痛、胃肠道刺激、血液毒性和肝脏毒性等。Roos等[76]用人肝癌细胞系HepG-2和HuH-7细胞研究了伊马替尼导致肝毒性的机制。他们发现在给予伊马替尼后,细胞中的溶酶体数量和体积都显著增加,通过吖啶橙染色评估溶酶体pH,结果显示伊马替尼升高了溶酶体pH并扰乱了水解酶的活性。溶酶体功能的障碍激活了转录因子TFEB,TEFB具有增加溶酶体合成和增强自噬的功能。对自噬相关基因BECLIN1,P62,UVRAG和微管相关蛋白1轻链3β(microtubule associated protein 1 light chain 3 beta,MAP1LC3B)的表达进行检测发现伊马替尼上调了上述基因,且电子显微镜下观察可看到细胞中自噬体数量的增多。由于mTORC1和TEFB之间存在相关性,Roos等又对mTORC1进行了评估发现其含量下调。总体而言,伊马替尼会诱导溶酶体功能障碍,这导致TEFB的活化和mTORC1活性下降,细胞自噬增强,自噬体积累,并最终诱导细胞凋亡,从而产生了肝脏毒性。
5 基于自噬水平调控的抗肿瘤药物毒性干预策略
调控抗肿瘤药物毒性作用引起的自噬,也就是在自噬发生的不同阶段进行干预从而恢复机体正常的自噬水平。总体可以分为两方面,一是升高自噬水平来缓解药物毒性,二是降低过度激活的自噬水平以减轻毒性作用。
5.1 升高自噬水平减轻药物毒性
某些抗肿瘤药物在发挥治疗作用的同时,可能会降低某些脏器细胞的自噬水平,导致一些有害蛋白或受损细胞器的积累,从而产生一定的毒性。因此,通过合适的途径升高细胞的自噬水平是一种可行的治疗方法。
抗癌抗生素博来霉素能通过抑制胸腺嘧啶核苷酸掺入DNA,从而干扰DNA的合成,在临床上用于治疗肺癌、宫颈癌、阴道癌、食道癌、头颈部及皮肤鳞状癌。然而,应用博来霉素的患者会出现间质性肺炎的症状,且常发展为肺纤维化,限制了博来霉素的应用和治疗效果[77]。在博来霉素诱导的小鼠肺纤维化模型中可观察到肺组织中LC3-Ⅱ/Ⅰ和BECLIN1的表达被显著抑制,而P62的表达增加,说明自噬受到抑制。先前的研究表明,博来霉素会引起PI3K/AKT/mTOR途径的异常激活,已有许多策略通过靶向此信号通路产生抗肺纤维化作用[78]。Mu等[79]通过生酮饮食模拟饥饿的代谢状态,抑制过度激活的PI3K/AKT/mTOR信号通路,促进自噬来减轻肺纤维化。此外,Baek等[80]研究发现采用内源性多胺——亚精胺,同样可以诱导自噬和抑制内质网应激来缓解博来霉素诱导的肺纤维化。
5.2 降低自噬水平减轻药物毒性
另一些抗肿瘤药物在治疗疾病时会导致自噬的过度激活,从而导致细胞中一些关键蛋白被降解,或是引起细胞程序性死亡而产生毒性作用,因此通过相应手段抑制自噬能够减轻毒性。
抗肿瘤抗生素阿霉素的抗瘤谱较广,对多种肿瘤疾病如白血病、恶性淋巴瘤和乳腺癌等具有治疗作用。然而阿霉素的心脏毒性是其最严重的毒性和副作用,限制了其在临床上的应用。目前提出的阿霉素引起心脏毒性的机制包括氧化应激增加、活性氧(reactive oxygen species,ROS)积累、心肌细胞钙离子水平失调以及细胞自噬和凋亡水平升高等[81]。 自噬在阿霉素的心脏毒性中起到一定的作用,阿霉素可能通过P53介导的GATA结合蛋白4(GATA binding protein 4,GATA4)抑制诱导自噬启动,导致BCL-2蛋白下调,BECLIN1,ATG5,ATG12和ATG4等蛋白上调,过度激活的自噬导致了心肌细胞死亡[82]。 在治疗策略上,研究表明二甲双胍能够降低细胞中H2O2的水平,并通过AMPK激活为线粒体损伤提供保护机制,其还能抑制自噬的异常激活,维持大鼠自噬标志物如LC3B-Ⅱ和P62在正常水平,从而改善心脏功能[83]。此外,还有研究表明姜黄素可上调PI3K/AKT/mTOR通路,减少自噬水平而显示出对心脏毒性的保护作用[84]。
中草药雷公藤的主要有效成分为雷公藤甲素(triptolide,TP),临床上广泛用于治疗各种肿瘤、炎症和自身免疫性疾病。然而,TP的毒性和副作用明显,对消化系统、生殖系统和泌尿系统均有损伤作用,其中肝脏毒性最为明显。TP导致肝毒性的机制十分复杂,包括脂质过氧化损伤、肝脏代谢紊乱、诱发自噬和凋亡等[85-86]。Zhang等[87]研究发现,给予TP后肝细胞中的LC3-Ⅱ和BECLIN1表达上调,表明其可激活自噬进程。TP合用mTOR抑制剂RAPA提高自噬水平会进一步降低肝细胞活力,合用自噬抑制剂3-MA降低自噬水平则能逆转毒性,证明自噬激活是TP诱导的肝细胞损伤的重要机制之一。先前有研究表明,梓醇对TP诱导的肝毒性具有保护作用,Zhang等[87]进一步研究发现TP诱导的过度自噬是通过蛋白激酶R样内质网激酶(protein kinase R-like endoplasmic reticulum kinase,PERK)-活化转录因子4(activating transcription factor 4,ATF4)-C/EBP同源蛋白(C/EBP-homologous protein,CHOP)通路介导的内质网应激导致的,而梓醇可以抑制PERK-ATF4-CHOP 通路,下调过度的自噬,从而达到缓解TP肝毒性的作用。
6 总结与展望
肿瘤是严重威胁人类健康的重要疾病之一,抗肿瘤药物的研发始终受到广泛关注。然而,抗肿瘤药物常存在安全性问题,主要集中于器官的毒性问题,这也是导致抗肿瘤药物研发失败的关键因素,因此寻找抗肿瘤药物毒性的干预策略成为一个亟待解决的问题。自噬是细胞中重要的物质降解途径,在许多生理病理活动中发挥作用。近年来随着人们对自噬机制的认识的不断深入,自噬和药物毒性之间的研究也逐渐增多。在抗肿瘤药物引发的脏器毒性中,自噬发挥的角色具有双向作用。一是抗肿瘤药物引发的毒性可激活细胞的保护性自噬,二是抗肿瘤药物引起细胞的致死性自噬,这可能与关键蛋白的降解或自噬体-溶酶体融合的阻断有关。针对基于自噬水平调控的干预策略来说,使用自噬泛抑制剂往往会抑制基础自噬水平,对心脏和肝脏等基础代谢水平较高的脏器造成损伤。因此更需对抗肿瘤药物引起过度自噬的机制进行深入研究,找到合适的干预手段,克服抗肿瘤药物毒性问题,同时这也有助于建立精准毒性筛选模型,提高药物研发成功率。
目前有关自噬与抗肿瘤药物毒性之间的研究主要集中于自噬下游关键底物蛋白的变化,如自噬的过度激活影响了心肌细胞中CCN2的表达水平,使得心肌细胞凋亡从而产生了心脏毒性。然而,在抗肿瘤药物毒性的发生和发展过程中起作用的可能有数种蛋白,仅研究某种底物蛋白的含量变化难以全面而准确地揭示抗肿瘤药物的毒性机制,并且仅对底物蛋白进行研究也难以推动毒性干预策略的发现。药物毒性的干预靶标通常是在某一信号通路的上游,然而目前针对抗肿瘤药物调控自噬水平的上游关键靶蛋白的研究还不够深入,尚不确定抗肿瘤药物是否通过影响AMPK,MAPK,PI3K/AKT/mTOR等上游激酶的活性引起自噬水平的改变。相应的研究有助于找到对应的干预策略,比如合用其他药物,在不影响抗肿瘤治疗效果的同时减少自噬相关的毒性和副作用。另外,明确上游位点参与调控自噬水平的关键作用也是自噬领域的研究重点之一,由于不同的抗肿瘤药物导致毒性的机制存在差异,以此为切入点找到对应的自噬相关上游位点并深入研究该位点参与自噬调控的普适性,可以发现自噬调控的全新分子机制,进一步促进自噬的基础理论研究,但仍需利用更多自噬评价模型去深入探索关键位点与自噬的整个进程之间的关系。
