厚朴酚固体分散体的制备及体外溶出试验
2022-02-08张雅倩赵兴华
张雅倩 , 曹 佳 , 赵兴华 , 何 欣
(河北农业大学动物医学院,河北 保定 071000)
厚朴酚(Magnolol,Mag)是从中药厚朴(Mangnoliaofficinalis)中提取出来的一种联苯酚类化合物[1]。厚朴酚具有抗炎、镇痛、抗氧化、抗肿瘤、抗病原微生物、降血糖和降血脂等作用,此外,厚朴酚还可发挥镇咳、抗腹泻、抗抑郁和降血压的疗效[2-4]。但由于厚朴酚在水中的溶解度只有11.6 μg/mL[5],导致其口服生物利用度差,大大限制了其临床应用。固体分散体(Solid dispersion,SD)制备方法简单,可有效提高药物溶解度从而显著提升药物的生物利用度。有报道以聚乙烯吡咯烷酮(PVP K30)为载体,采用溶剂挥发法制备欧前胡素固体分散体,欧前胡素的表观溶解度由原料药的11.82 μg/mL提升到179.09 μg/mL,累积溶出率高达94.26%,比物理混合物高出近3倍[6]。甲基丙烯酸-丙烯酸乙酯共聚物1∶1(Methacrylic acid ethyl acrylate copolymer,ME)属于肠溶性聚丙烯酸树脂类聚合物,其成膜性好,安全无毒,机体不吸收,主要用作片剂、微丸、缓释颗粒等固体缓控释制剂的薄膜包衣,具有性能稳定的优点[7],目前以ME用作固体分散体载体的报道相对较少。
本试验以厚朴酚为模型药物,以ME为载体材料,选用传统固体分散体制备方法——溶剂蒸发法,制备不同药物和载体比例的固体分散体,对固体分散体进行表征分析,并比较不同比例的固体分散体的平衡溶解度和累积溶出率,旨在提高厚朴酚的溶出性能,从而扩大厚朴酚的临床应用,为难溶性活性成分的开发提供参考思路。
1 材料与方法
1.1 主要药品和试剂 厚朴酚标准品(纯度≥99%,批号:110729),购自中国食品药品检定研究院;厚朴酚原料药(纯度≥98%,批号:RL20210312),购自西安瑞林生物科技有限公司;甲基丙烯酸-丙烯酸乙酯共聚物(1∶1)粉末,购自北京凤礼精求医药股份有限公司;磷酸二氢钠和磷酸二氢钠,均购自上海阿拉丁试剂有限公司;无水乙醇(分析纯),购自天津进丰化工有限公司。
1.2 主要仪器 BSA124S电子分析天平,北京赛多利斯科学仪器有限公司产品;RE-52AA旋转蒸发仪,上海亚荣生化仪器厂产品;79-1型磁力搅拌器,北京中兴伟业仪器有限公司产品;LGJ-18型冷冻干燥机,北京松源华兴科技发展有限公司产品;Q2000差示扫描量热仪,上海METTLER TOLEDO产品;TD-3700型X射线衍射仪,丹东通达科技有限公司产品;6850UV紫外-分光光度计,美国JENWAY产品;Nicolet Avatar 370傅立叶变换红外光谱仪,德国BRUKER产品;ZEISS MERLIN Compact电镜扫描仪,德国卡尔察司公司产品;BY-211C卧式空气浴摇床,上海BAJIU产品;PES 0.45 μm水系针头滤器,天津津腾实验设备有限公司产品。
1.3 固体分散体的制备 精密称取厚朴酚原料药和ME,按1∶3、1∶5、1∶7、1∶9 的质量比例混匀,加入适量无水乙醇,充分溶解后移至梨形瓶,旋转蒸发并收集剩余物质,冷冻干燥后,研磨过筛,在真空干燥器中密封保存备用。
1.4 厚朴酚测定方法的建立 取厚朴酚标准品适量,以无水乙醇稀释,配制成系列浓度(20、10、5、2.5、1.25 μg/mL和0.625 μg/mL)的标准曲线工作液,用紫外可见分光光度计在290 nm处测定各个浓度下厚朴酚的吸光度。用Origin 2017软件对测得的厚朴酚的吸光度(A)和已知标准浓度(c)做线性回归。
1.5 固体分散体的表征
1.5.1 粉末X射线衍射(PXRD) 分别取5~10 mg各质量比例的厚朴酚固体分散体、厚朴酚原料药、载体ME以及物理混合物(Physical mixture,PM),用X射线衍射仪进行检测。设置条件:光源为Cu Kα(波长1.5418Å),电压和电流分别设置为30 kV和20 mA,2θ的扫描范围为5~35°,测试步宽为0.02°,扫描速度为每步0.25 s,绘制X射线衍射图。
1.5.2 差示扫描量热(DSC) 取3~5 mg各质量比例的厚朴酚固体分散体、厚朴酚原料药、载体ME以及物理混合物置于铝坩埚内,将空铝坩埚作为样品对照,用差示扫描量热仪检测。测定条件:升温速度为10.0 K/min,温度扫描范围为25~200 ℃,氮气流速为50 mL/min,绘制DSC图。
1.5.3 傅里叶红外光谱(FT-IR) 取5~10 mg质量比例为1∶5的厚朴酚固体分散体、厚朴酚原料药、载体ME以及质量比例为1∶5的物理混合物,置于红外光谱仪中测试。设定条件:横坐标波数的扫描范围为4 000~400 cm-1,分辨率为0.1 cm-1,记录相应谱图。
1.5.4 扫描电子显微镜(SEM) 取5~10 mg质量比例为1∶5的厚朴酚固体分散体、厚朴酚原料药、载体ME以及质量比例为1∶5的物理混合物,用扫描电镜进行观察。测试条件:仪器选用ZEISS MERLIN Compact 扫描电子显微镜,电压设置为20 kV,样品置于铜板上并用导电胶带固定,并用金喷射。
1.6 最佳载药比例的确定
1.6.1 固体分散体平衡溶解度的测定 采用经典摇瓶法测定平衡溶解度[8]。平行称取3份过量的各质量比例的厚朴酚固体分散体和厚朴酚原料药,分别置于6 mL pH 6.8磷酸缓冲液中,密封置于37 ℃的恒温振荡仪中,以100 r/min振荡24 h,取出,经0.45 μm的水性滤膜过滤,过滤后的液体适当稀释后,经UV法测定药物的吸光度,代入标准曲线,以所得的药物浓度计算药物的平衡溶解度。
1.6.2 固体分散体累积溶出率的测定 选用200 mL pH 6.8磷酸缓冲液为溶出介质,设置温度为(37.0±0.5)℃,转速为250 r/min。平行称取3份过量的厚朴酚固体分散体和厚朴酚原料药分别置于溶出介质中,并同时加入10 μL吐温80,自样品与介质接触开始计时,于0.55、10、20、30、45、60、90、120、180 min和240 min取样500 μL,并补充等温等体积介质。取出的样品经0.45 μm的水性滤膜过滤,适当稀释后,经UV法测定吸光度,代入标准曲线,得到药物浓度并计算累积溶出率:
其中:Q为累积溶出率;Ci和Ci-1为各时间点取出的样品浓度;V取样为各时间点固定取样体积;V溶出介质为溶出介质体积。
2 结果
2.1 厚朴酚测定方法的建立 测定得到厚朴酚原料药的线性回归方程:A=0.026 59c+0.007 71,r2=0.999 96;表明厚朴酚在0.625~20 μg/mL浓度范围内线性关系良好。
2.2 固体分散体的表征
2.2.1 粉末X射线衍射 如图1所示,厚朴酚原料药的X射线衍射图中有强烈的衍射峰,峰的位置分别在11.38°、13.30°、17.02°、19.92°、21.82°和26.82°处。物理混合物存在与原料药相同位置的晶体衍射峰,表明药物与载体只是简单混合。本试验所制备厚朴酚固体分散体的PXRD图谱显示,原料药的晶体特征衍射峰全部消失,说明该固体分散体中厚朴酚在体系中以非晶体状态分散于载体中,形成无定形固体分散体。
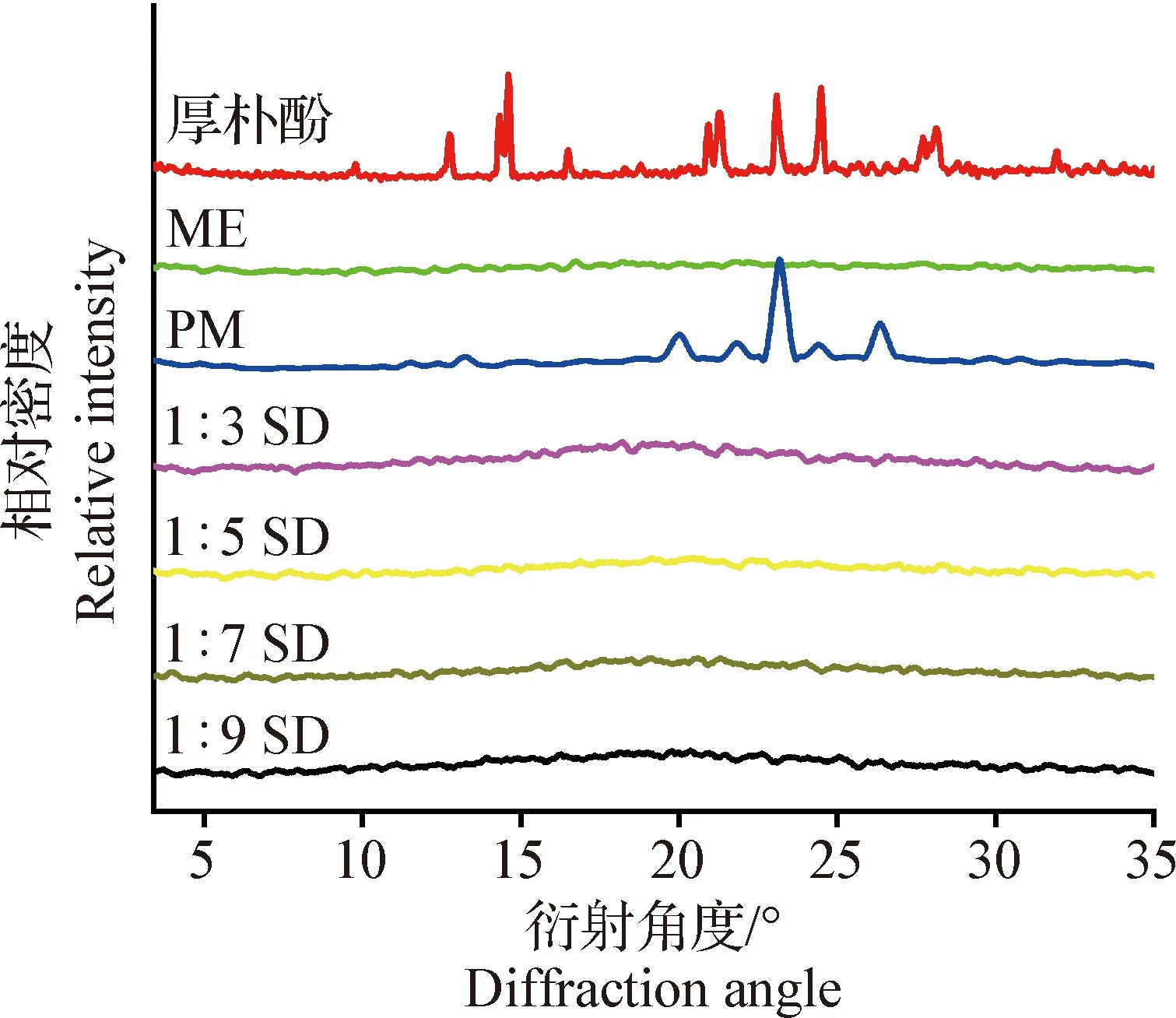
图1 厚朴酚、ME、物理混合物和固体分散体的PXRDFig.1 PXRD of magnolol,ME,physical mixture and solid dispersion
2.2.2 差示扫描量热 如图2所示,厚朴酚在102 ℃左右有1个尖锐的吸热峰,与厚朴酚原料药的熔点一致[9];载体ME在25~200 ℃温度范围内未产生吸热峰;物理混合物和厚朴酚原料药一样,在102 ℃左右具有相同的特征吸热峰,表明物理混合物并没有改变厚朴酚的晶型状态,二者只是简单的物理混合;而制备的厚朴酚固体分散体在温度扫描范围内(25~200 ℃)并未出现晶体吸热峰,说明厚朴酚是以无定形态分散在聚合物中。
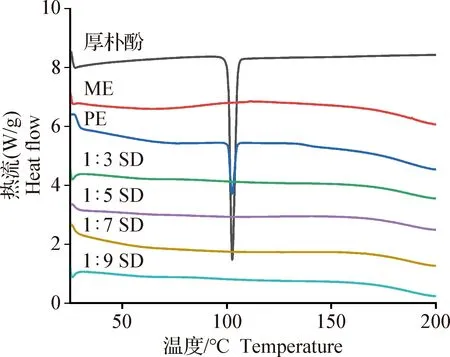
图2 厚朴酚、ME、物理混合物和固体分散体的DSCFig.2 DSC of magnolol,ME,physical mixture and solid dispersion

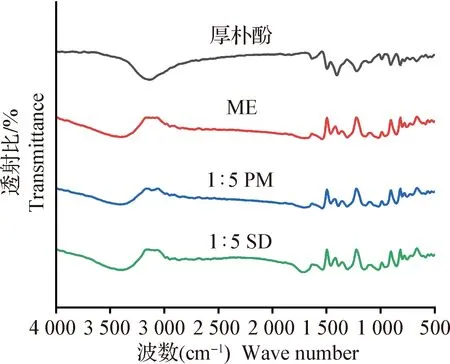
图3 厚朴酚、ME、物理混合物和固体分散体的FT-IRFig.3 FT-IR of magnolol,ME,physical mixture and solid dispersion
2.2.4 扫描电镜分析 电镜扫描结果如图4所示,厚朴酚为大小不一的块状晶体;载体ME为表面凹凸不平的球状颗粒;物理混合物的电镜扫描图像可以很明显地看到厚朴酚和ME只是简单混合,既有块状晶体,又有球状颗粒;1∶5 SD中几乎不存在明显的晶体,表明厚朴酚均匀分散于载体中,形成了一种相对均一的物质,与DSC和PXRD的结果一致。
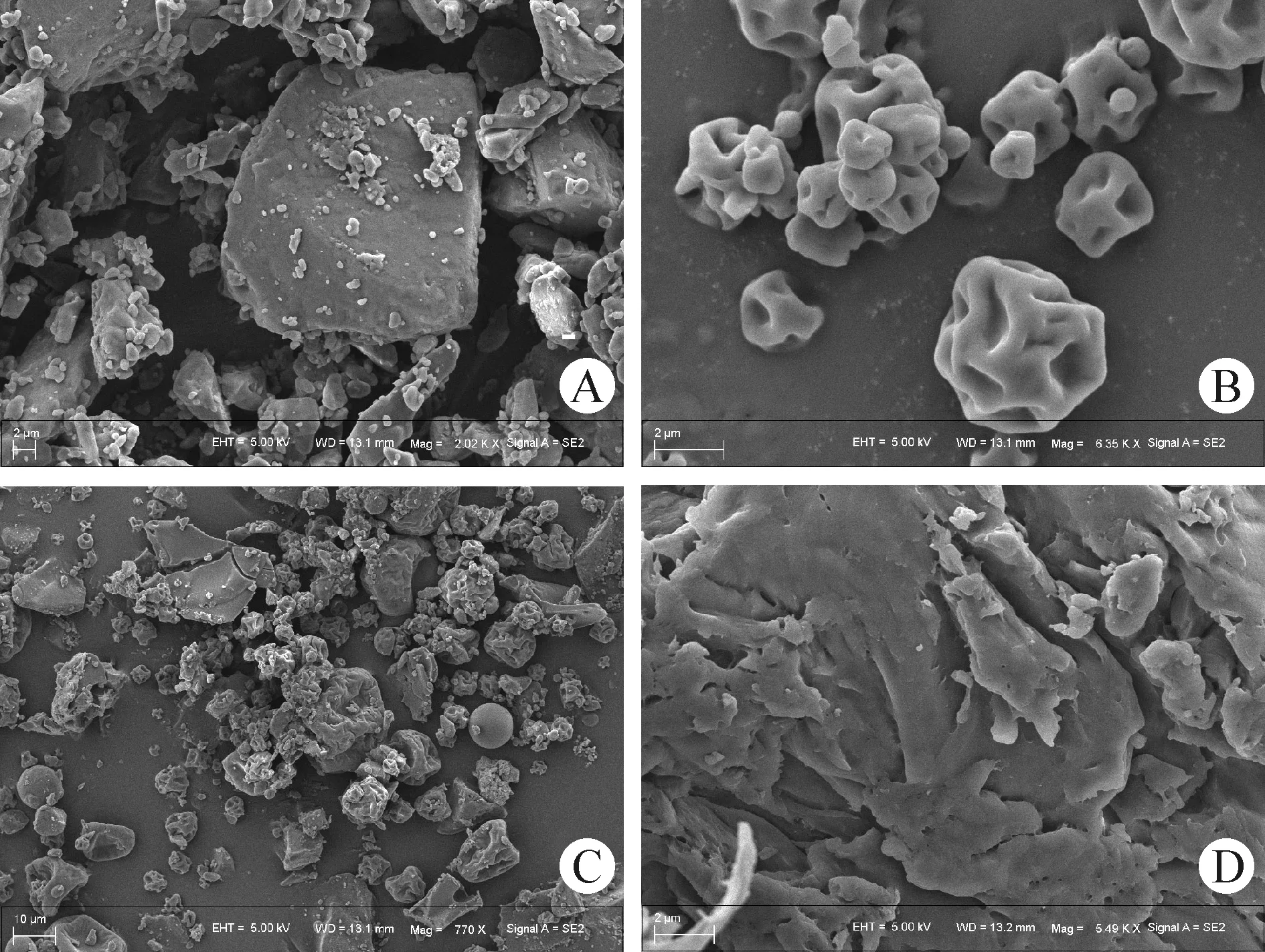
图4 厚朴酚、ME、物理混合物和固体分散体的SEMFig.4 SEM of magnolol,ME,physical mixture and solid dispersion A:厚朴酚原料药(2 020×); B:ME(6 350×); C:1∶5物理混合物(770×); D:1∶5固体分散体(5 490×)A:Magnolol raw material medicine(2 020×); B:ME(6 350×); C:1∶5 physical mixture(770×); D:1∶5 solid dispersion(5 490×)
2.3 最佳载药比例的确定
2.3.1 固体分散体平衡溶解度的测定 如表1所示,固体分散体均能够提高厚朴酚在pH 6.8磷酸缓冲液中的平衡溶解度,其中质量比例为1∶5、1∶7和1∶9的厚朴酚固体分散体可以显著提升厚朴酚的平衡溶解度达5倍以上。
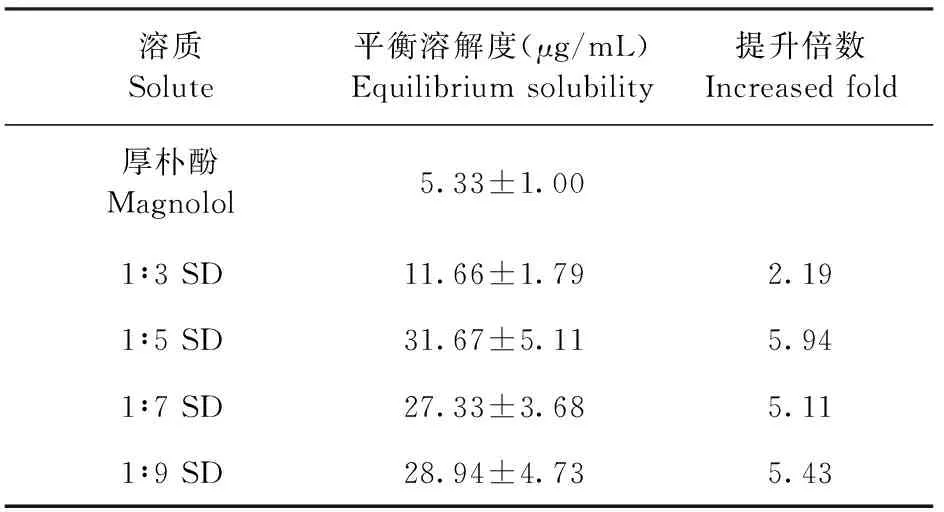
表1 厚朴酚原料药和厚朴酚固体分散体在pH 6.8磷酸缓冲液中的平衡溶解度Table 1 Equilibrium solubility of magnolol raw materials and magnolol solid dispersions in pH 6.8 phosphate buffer
2.3.2 固体分散体累积溶出率的测定 如图5所示,3个比例的厚朴酚固体分散体在4 h内均能有效提高厚朴酚的累积溶出率。厚朴酚在4 h内的累积溶出率为36.74%,1∶5 SD、1∶7 SD分别达90.56%、83.56%,分别是原料药的2.46、2.27倍,1∶9 SD在4 h内的累积溶出率最高,为97.28%,为原料药的2.65倍。其中1∶9 SD和1∶7 SD达平衡浓度较快,1∶5 SD略显缓慢。
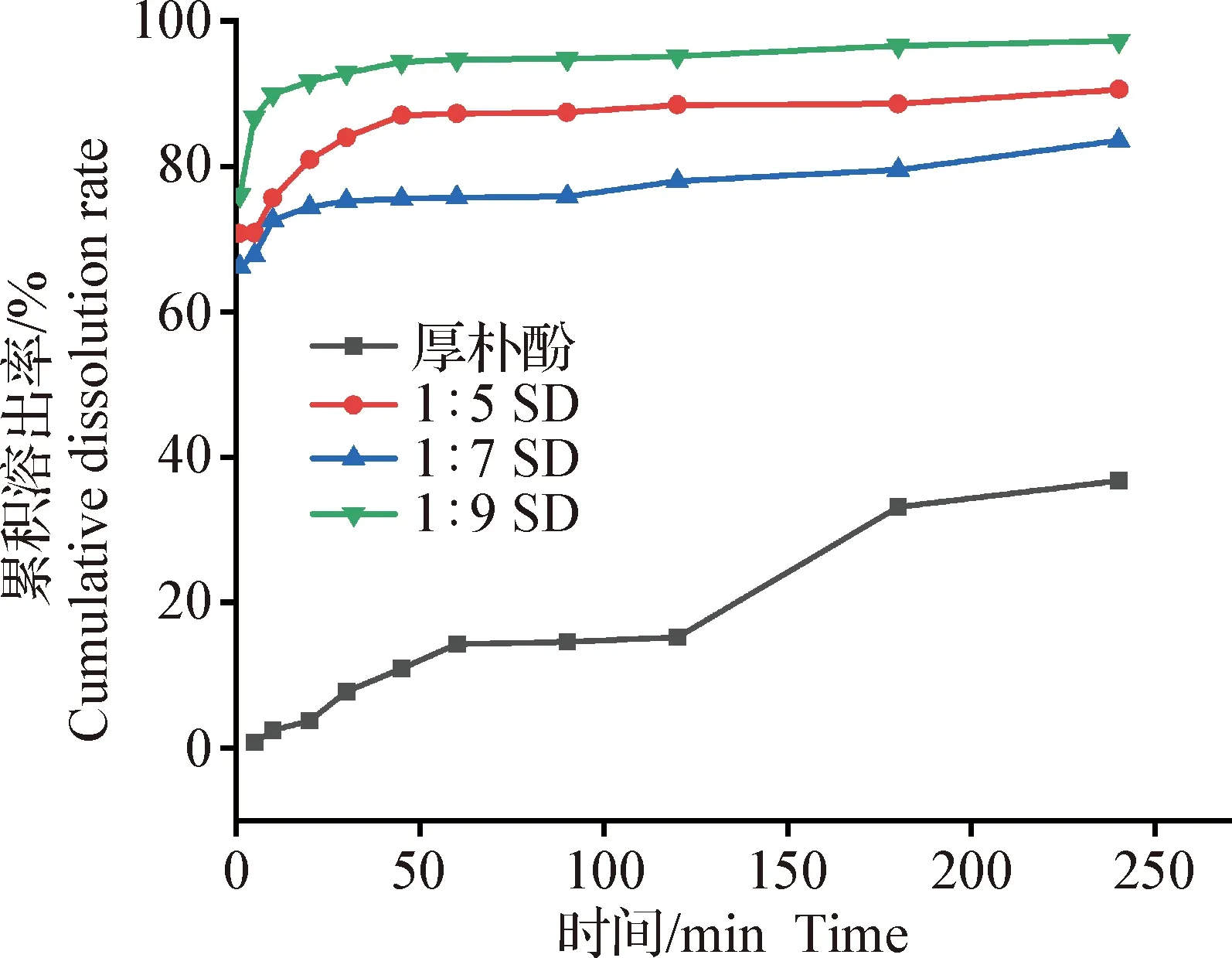
图5 不同载药比例厚朴酚固体分散体的溶出曲线Fig.5 Dissolution curves of magnolol solid dispersions with different drug loading ratios
3 讨论
厚朴酚为难溶性药物,口服生物利用度只有4.9%[10],在临床应用中大大受限。关于提升厚朴酚溶解度的文献已十分广泛,所制成的制剂类型也多种多样,目前已经研制出的厚朴酚制剂包括固体分散体、磷脂复合物、纳米晶体混悬剂、脂质体、纳米粒等[11-15],在提升厚朴酚的溶解度方面,这几种制剂都取得了不错的效果。考虑到制备工艺的复杂程度和有效性,本试验选用了工艺相对简单但有效的固体分散体作为厚朴酚的制剂类型。
在固体分散体中,药物以非晶体的形式存在,理论上,非晶药物的自由能比晶体药物高,溶出的过程中不需要消耗额外的动力克服晶格能,故而具有更高的溶解度和溶出度[16]。固体分散体技术目前已经广泛应用于难溶性中药提取物溶解度的提升,吕志阳等[17]采用热熔挤出技术制备了银杏总内酯-聚丙烯酸树酯EPO固体分散体,该固体分散体对银杏总内酯有很好的抑晶作用,使得累积溶出率由物理混合物的70%提升至固体分散体的90%;张守徳等[18]选用聚丙烯酸树脂II为载体,使用喷雾干燥法成功制备穿心莲内酯固体分散体,改善了固体分散体稳定性差的情况,并提高了药物润湿性,使得其累积溶出率接近100%,较原料药提高5倍左右。上述研究与本试验的相同之处在于目标药物都属于中药提取物,载体选用类别都属于聚丙烯酸树脂类药物,在制备方法方面有所不同,热熔挤出技术对温度有要求,通常需要在高温下进行操作,如果温度控制不当会导致药物降解,影响药物的稳定性等问题,因此该方法对药物有温度的要求;喷雾干燥法需要机器配置的支持,且配方变量方面的要求等会影响到药物的干燥效率,进而影响固体分散体的固态性能;这2种方法在制备方面都有所局限,而本试验采用的是溶剂挥发法,无溶剂残留问题,适用于遇热不稳定的药物,并且药物在载体中的分散性较好,溶出速率较快[19-21]。
本试验以ME为载体,采用溶剂蒸发法成功制备了厚朴酚固体分散体,并筛选出厚朴酚固体分散体最优制备比例为1∶9。ME作为固体缓控释剂的包衣材料,具有性能稳定、成膜性好的优点,能够有效包被厚朴酚。此外,溶剂蒸发法只是将溶剂蒸出,载体与药物几乎被完整保留下来,从而避免了药物浪费,提升了出品率。厚朴酚固体分散体溶解度提高的主要原因是药物在载体中高度分散成无定形态,能对药物产生良好的润湿效果,因此溶解性提高。红外结果显示厚朴酚固体分散体中可能产生了分子间氢键,该氢键使得厚朴酚固体分散体在溶剂中容易被水包围,有助于药物的分散,为累积溶出率的提升提供了一定依据。
