HPLC法同时测定强力定眩片中13种成分
2022-01-27罗欢欢冯泽宇谢艳华田慧玲肖会敏王四旺李志平
罗欢欢, 冯泽宇, 李 捷, 谢艳华, 田慧玲, 肖会敏, 王四旺*, 李志平
(1.西北大学生命科学与医学部,陕西 西安 710069;2.陕西汉王药业有限公司,陕西 汉中 723000;3.空军军医大学中药与天然药物教研室,陕西 西安 710032)
强力定眩片是陕西汉王药业有限公司独家生产的复方制剂,由天麻、杜仲、杜仲叶、野菊花、川芎组成,主治高血压、高脂血症、眩晕,临床疗效显著[1-4]。中药(尤其是复方)组成复杂,多成分定量分析能更全面客观地评价其质量[5-7],但文献[8-12]仅以天麻素为强力定眩片质控指标,只有王媚[13]采用UPLC-ESI-MS/MS法同时测定其中7种成分含量。
2015、2020年版《中国药典》将天麻素和对羟基苯甲醇、松脂醇二葡萄糖苷、绿原酸、蒙花苷分别作为天麻、杜仲、杜仲叶、野菊花指标成分,藁本内酯具有抗动脉粥样硬化、抗脑缺血等药理作用[14],没食子酸具有降血压作用[15],5-羟甲基糠醛具有改善学习记忆、保护神经细胞等药理作用[16],绿原酸具有保护心血管作用[17-18]。本实验建立HPLC法同时测定强力定眩片中天麻素、没食子酸、5-羟甲基糠醛、对羟基苯甲醇、新绿原酸、绿原酸、隐绿原酸、巴利森苷B、松脂醇二葡萄糖苷、巴利森苷C、巴利森苷A、藁本内酯、蒙花苷的含量,以期为该制剂质量控制提供理论和技术支持。
1 材料
1.1 仪器 LC-2010CHT高效液相色谱仪(日本岛津公司);BSA2202S型电子天平(德国Sartorius公司);移液枪(德国Eppendorf公司);Milli-Q Integral 5超纯水一体化系统(美国密理博公司);KQ-500E超声清洗器(昆山市超声仪器有限公司)。
1.2 试剂与药物 强力定眩片10批,批号(编号)分别为017101(S1)、024102(S2)、016101(S3)、023102(S4)、027102(S5)、019101(S6)、021101(S7)、026102(S8)、028102(S9)、007101(S10),由陕西汉王药业股份有限公司提供。天麻素(批号HR16313B1,纯度≥98.0%)、5-羟甲基糠醛(批号HH248894198,纯度≥98.0%)、对羟基苯甲醇(批号H21D6Q7813,纯度≥98.0%)、新绿原酸(批号HR20422B1,纯度≥98.0%)、隐绿原酸(批号HR20423B1,纯度≥98.0%)、巴利森苷B(批号HR2098B1,纯度≥98.0%)巴利森苷C(批号HS20905B1,纯度≥98.0%),巴利森苷A(批号HS11129W4,纯度≥98.0%)、松脂醇二葡萄糖苷(批号HS0870W2,纯度≥98.0%)、藁本内酯(批号HL09355198,纯度≥98.0%)对照品均购自宝鸡辰光生物科技有限公司;绿原酸(批号110753-201415,纯度≥96.20%)、蒙花苷(批号111528-201300,纯度≥95.1%)、没食子酸(批号110831-201906,纯度≥91.5%)对照品均购自中国食品药品检定研究院。乙腈、甲醇均为色谱纯,购自美国Fisher公司;其他试剂均为分析纯;水为超纯水。
2 方法与结果
2.1 色谱条件 Lamdo Stamsil C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈(A)-0.05%磷酸(B),梯度洗脱(0~2 min,3%~7%A;2~20 min,7%A;20~30 min,7%~12%A;30~90 min,12%~30%A;90~100 min,30%~50%A);体积流量0.7 mL/min;柱温20 ℃;检测波长220、280 nm;进样量5 μL。
2.2 供试品溶液制备 取本品10片,去薄膜衣,研细,精密称取约0.5 g,置于具塞锥形瓶中,精密加入50 mL 50%甲醇,密塞,称定质量,超声(功率250 W、频率40 kHz)处理30 min,放冷,50%甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.3 对照品溶液制备 精密称取各对照品适量,甲醇制成每1 mL分别含天麻素212.50 μg、没食子酸233.00 μg、5-羟甲基糠醛136.50 μg、对羟基苯甲醇208.00 μg、新绿原酸493.00 μg、绿原酸224.50 μg、隐绿原酸227.50 μg、巴利森苷B 125.00 μg、松脂醇二葡萄糖苷122.00 μg、巴利森苷C 160.50 μg、巴利森苷A 154.00 μg、藁本内酯2 225.00 μg、蒙花苷234 μg的溶液,即得。
2.4 阴性样品溶液制备 按照本品处方,分别制得缺天麻、缺杜仲、缺杜仲叶、缺野菊花、缺川芎的阴性样品,按“2.2”项下方法制备,即得。
2.5 专属性试验 精密吸取供试品、对照品、阴性样品溶液,在“2.1”项色谱条件下进样测定,结果见图1。由此可知,对照品、供试品溶液在相应位置处有相同色谱峰,各成分色谱峰分离度均大于1.5,阴性无干扰,表明该方法专属性良好。
2.6 线性关系考察 精密吸取“2.3”项下对照品溶液适量,稀释成6个质量浓度,在“2.1”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,结果见表1,可知各成分在各自范围内线性关系良好。
2.7 精密度试验 精密吸取“2.2”项下供试品溶液,在“2.1”项色谱条件下进样测定6次,测得天麻素、没食子酸、5-羟甲基糠醛、对羟基苯甲醇、新绿原酸、绿原酸、隐绿原酸、巴利森苷B、松脂醇二葡萄糖苷、巴利森苷C、巴利森苷A、藁本内酯、蒙花苷峰面积RSD分别为2.03%、1.05%、1.21%、1.15%、1.26%、1.66%、0.70%、1.32%、1.29%、0.98%、1.91%、1.94%、1.64%,表明仪器精密度良好。
2.8 重复性试验 称取本品(批号007101)6份,按“2.2”项下方法制备供试品溶液,在“2.1”项色谱条件进样下测定,测得天麻素、没食子酸、5-羟甲基糠醛、对羟基苯甲醇、新绿原酸、绿原酸、隐绿原酸、巴利森苷B、松脂醇二葡萄糖苷、巴利森苷C、巴利森苷A、藁本内酯、蒙花苷峰面积RSD分别为1.26%、1.62%、1.90%、1.90%、1.27%、2.12%、1.80%、2.20%、2.11%、1.70% 1.99%、1.60%、1.68%,表明该方法重复性良好。
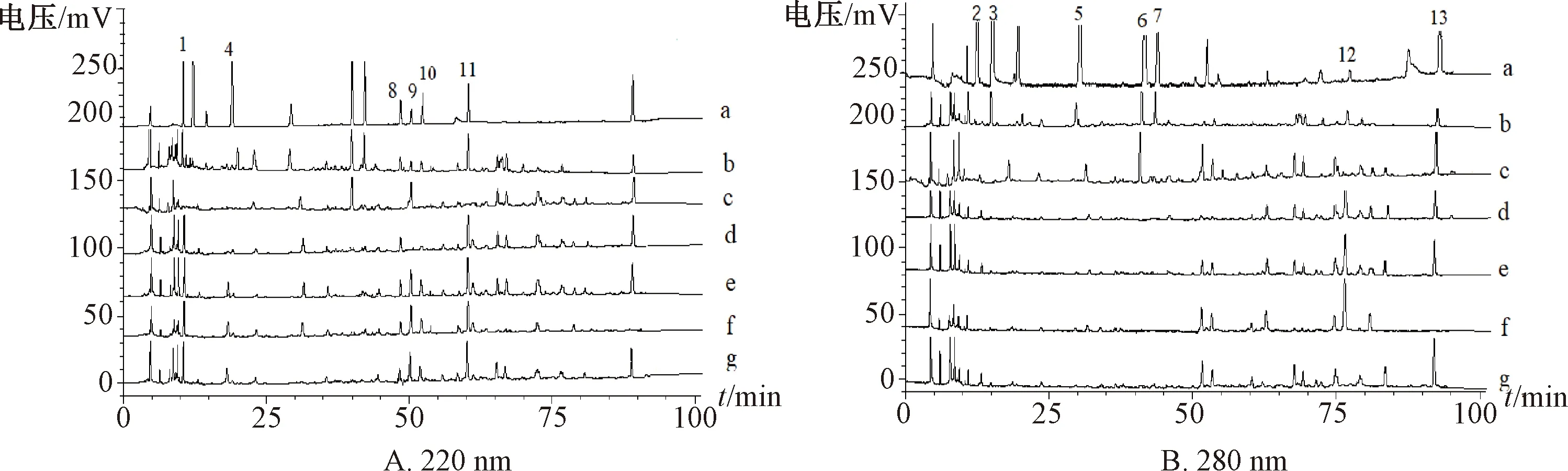
注:a~g分别为对照品、供试品、缺天麻阴性样品、缺杜仲阴性样品、缺杜仲叶阴性样品、缺野菊花阴性样品、缺川芎阴性样品。1.天麻素 2.没食子酸 3.5-羟甲基糠醛 4.对羟基苯甲醇 5.新绿原酸 6.绿原酸 7.隐绿原酸 8.巴利森苷B9.松脂醇二葡萄糖苷 10. 巴利森苷C 11. 巴利森苷A 12.藁本内酯 13.蒙花苷1.gastrodia elata 2.gallic acid 3.5-hydroxymethyl furfural 4. para-hydroxybenzyl alcohol 5. neochlorogenic acid 6.chlorogenic acid 7. cryptochlorogenic acid 8. parisin B 9. pinolinol diglucoside 10. parisin C 11. parisin A 12. ligustilide 13. linarin图1 各成分HPLC色谱图Fig.1 HPLC chromatograms of various constituents

表1 各成分线性关系
2.9 稳定性试验 精密吸取“2.2”项下供试品溶液适量,于0、2、4、6、8、10、12、24、48 h在“2.1”项色谱条件下进样测定,测得天麻素、没食子酸、5-羟甲基糠醛、对羟基苯甲醇、新绿原酸、绿原酸、隐绿原酸、巴利森苷B、松脂醇二葡萄糖苷、巴利森苷C、巴利森苷A、藁本内酯、蒙花苷峰面积RSD分别为2.01%、1.56%、1.55%、1.08%、2.24%、2.29%、1.20%、1.31%、1.99%、2.00%、2.30%、2.96%、2.42%,表明溶液在48 h内稳定性良好。
2.10 加样回收率试验 取本品(批号007101)约0.25 g,共6份,精密称定,精密加入对照品溶液,按“2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,计算回收率。结果,天麻素、没食子酸、5-羟甲基糠醛、对羟基苯甲醇、新绿原酸、绿原酸、隐绿原酸、巴利森苷B、松脂醇二葡萄糖苷、巴利森苷C、巴利森苷A、藁本内酯、蒙花苷平均加样回收率分别为102.43%、104.79%、97.91%、99.43%、104.99%、101.66%、102.75%、97.34%、98.64%、100.70%、100.79%、105.29%、102.69%,RSD分别为2.07%、0.70%、1.97%、2.34%、0.64%、2.26%、2.22%、0.67%、1.70%、1.96%、2.88%、2.62%、1.49%。
2.11 样品含量测定 取本品10批,按“2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,计算含量,结果见表2。
2.12 聚类分析 采用SPSS 26.0软件中的组间连接聚类,以欧氏距离为测度,对10批样品进行聚类分析,结果见图2。由此可知,当类间距离为10时,10批样品可聚为2类,S3、S7、S5、S1、S8、S6为第Ι类,S4、S10、S2、S9为第Ⅱ类,可能与原药材产地、生长年限、采收季节、加工炮制方式等有关。
2.13 主成分分析 采用SPSS 26.0软件对10批样品进行主成分分析,以累积方差贡献率>85%、初始特征值>1为提取标准,发现前3个主成分初始特征值分别为6.673、2.632、2.038,均大于1;方差贡献率分别为51.331%、20.244%、15.674%,总贡献率为87.249%,大于85%,碎石图见图3,载荷矩阵见表3。由此可知,巴利森苷C、新绿原酸、隐绿原酸、绿原酸、蒙花苷、巴利森苷B、对羟基苯甲醇、巴利森苷A对主成分1贡献率高,没食子酸、5-羟甲基糠醛、天麻素、藁本内酯对主成分2贡献率高,松脂醇二葡萄糖苷对主成分3贡献率高。

表2 各成分含量测定结果(mg/g,n=3)
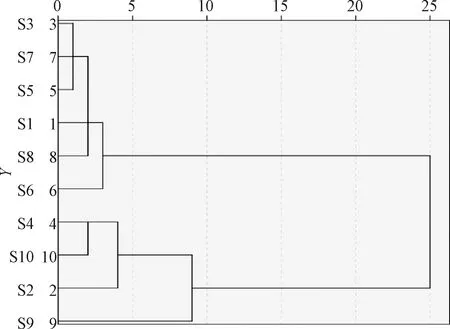
图2 10批样品聚类分析图Fig.2 Cluster analysis diagram for ten batches of samples

图3 各成分碎石图Fig.3 Scree plot for various constituents
2.14 质量评价 分别以F1、F2、F3代表3种主成分,作为10批样品所含13种成分(X1~X13)的信息,并建立线性表达式,即F1=0.13X1+0.147X2-0.135X3+0.314X4+0.357X5+0.336X6+0.355X7+0.314X8+0.244X9+0.377X10+0.26X11+0.077X12-0.324X13、F2=0.396X1+0.534X2+0.488X3-0.207X4+0.087X5+0.148X6+0.105X7-0.185X8-0.19X9-0.074X10-0.24X11+0.268X12-0.177X13、F3=0.268X1+0.043X2+0.273X3-0.228X4-0.245X5-0.264X6-0.24X7+0.35X8+0.485X9+0.067X10+0.412X11+0.238X12+0.155X13,将其与所对应主成分的方差贡献率相乘后,再除以累积贡献率,计算综合评分Y,表达式为Y=(F1×51.331+F2×20.244+F3×15.674)/87.249,结果见表4。由此可知,样品S5(批号027102)质量最优,其次是S3(批号016101)、S7(批号021101),S2(批号024102)、S9(批号028102)质量较差。

表3 初始因子载荷矩阵
3 讨论
3.1 色谱条件筛选 本实验以各成分分离度、保留时间、峰形为指标,比较了不同流动相[甲醇-水、乙腈-水、甲醇-水(含1%磷酸)、甲醇-水(含0.05%磷酸)、乙腈-水(含1%磷酸)、乙腈-0.05%磷酸、甲醇-水(含1%乙酸)、甲醇-水(含0.05%乙酸)]、色谱柱(Inertsil C18、Kromasil 100-5C18、Diamonsil C18、Lamdo Stamsil C18)、柱温(20、25、30、35 ℃)、体积流量(0.6、0.7、0.8、1.0 mL /min),最终分别确定为乙腈-0.05%磷酸、Lamdo Stamsil C18色谱柱、柱温20 ℃、体积流量0.7 mL/min。在190~800 nm范围内进行全波长扫描,发现天麻素、对羟基苯甲醇和巴利森苷B、巴利森苷C和巴利森苷A最大吸收波长分别为221、223、222 nm,故在220 nm下检测;没食子酸、5-羟甲基糠醛、藁本内酯、蒙花苷、新绿原酸、绿原酸、隐绿原酸最大吸收波长分别为272、283、277、333、324、324、324 nm,但新绿原酸、绿原酸、隐绿原酸在324 nm处峰形较差,而上述7种成分在280 nm处均有较好的灵敏度,杂质峰干扰较小,故选择其作为检测波长;松脂醇二葡萄糖苷在220 nm波长处的峰型优于280 nm波长处,故确定为220 nm。
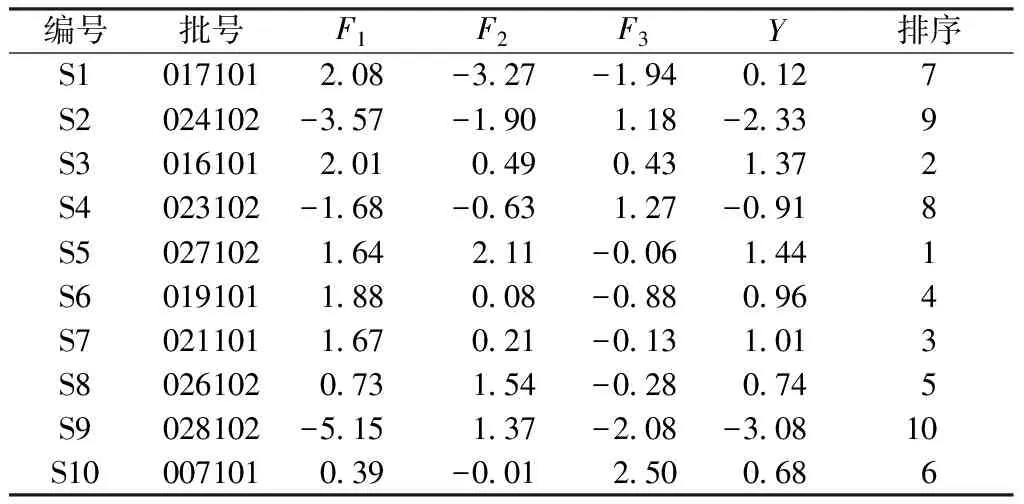
表4 10批样品综合评分
3.2 供试品溶液制备方法筛选 本实验首先考察了提取溶剂水、甲醇、乙醇,发现甲醇提取效果较好,再考察了体积分数50%、60%、70%、80%、90%,发现50%时提取率较高,峰面积较大,各成分色谱峰与杂质均能达到基线分离。然后,考察了加热回流、超声提取,发现藁本内酯等成分受热不稳定,故选择超声提取[19-20]。最后,考察了提取时间15、30、45 min,发现提取30 min时提取率高于提取15 min时,而提取30、45 min时无明显区别,故确定为30 min。
4 结论
本实验建立了HPLC法同时测定强力定眩片中天麻素、没食子酸、5-羟甲基糠醛、对羟基苯甲醇、新绿原酸、绿原酸、隐绿原酸、巴利森苷B、松脂醇二葡萄糖苷、巴利森苷C、巴利森苷A、藁本内酯、蒙花苷的含量,该方法快速准确,高效灵敏,专属性强,可为该制剂质量控制提供理论依据。
