抗氧化干预对低氧低糖诱导血管平滑肌细胞自噬和铁死亡的影响
2022-01-25杨光明彭小勇薛明英刘良明
杨光明,彭小勇,雷 艳,胡 弋,薛明英,李 涛,刘良明
(1.陆军特色医学中心野战外科研究部武器杀伤生物效应评估研究室,重庆 400042;2.陆军特色医学中心野战外科研究部战伤休克与输血研究室/创伤、烧伤与复合伤国家重点实验室,重庆 400042)
缺血缺氧是多种临床疾病中广泛存在的病理生理过程,是引起重要组织器官损伤的最常见因素之一。机体供血不足导致的组织缺血缺氧是发生严重创伤失血休克的最主要原因,其不仅会引起组织器官的直接损伤,还可能增加机体对细菌、毒素、炎症介质等因素的敏感性,在多种继发性损伤的发生发展中起重要作用[1-2]。因此,深入研究组织细胞缺血缺氧损伤的发生机理和调节过程对严重创伤失血休克等临床重症的治疗具有重要意义。
缺血缺氧可导致多种细胞应激反应的激活,如细胞自噬和铁死亡。自噬通过溶酶体系统对细胞自身组成成分进行降解并循环利用,在生理和病理状态下对细胞命运有着决定作用[3-4]。多项对缺血性疾病的研究显示,自噬增强可对缺血缺氧状态下的心肌细胞和神经元起保护作用,能够促进细胞存活。然而也有研究表明,自噬可通过自身介导的死亡机制或触发细胞凋亡等死亡通路,加重缺血缺氧后的细胞损伤程度,甚至诱导细胞死亡[5-7]。这些不同的结论与自噬自身“双刃剑”的特点有关,但自噬在缺血缺氧损伤中的作用及其调节机制仍未完全阐明。铁死亡是近年来发现的一种新的细胞死亡模式,其在肿瘤和神经等疾病中的重要作用已得到证实,近年来在出血性和缺血性疾病中也发现了铁死亡的参与[8-10]。然而,目前关于铁死亡的研究尚处于初期阶段,铁死亡的调节机制和临床作用尚需进一步探索。
氧化应激与自噬、铁死亡紧密相关。氧化应激激活的保护性自噬反应和氧化应激引起的自噬性细胞死亡均有报道[11-12]。而铁死亡以铁依赖的脂质过氧化损伤为主要特征,其可导致活性氧蓄积、细胞损伤死亡,与氧化应激密切相关。本实验室前期研究证实,氧化应激在创伤失血休克后血管功能障碍的发生过程中具有重要作用[13]。但目前尚不清楚在创伤失血休克后缺血缺氧状态下,氧化应激与自噬和铁死亡是否存在联系,亦不清楚调控氧化状态能否影响细胞自噬状态或铁死亡发生进而参与血管功能调节。为此,本研究采用体外培养的大鼠血管平滑肌细胞(vascular smooth muscle cells,VSMC),观察抗氧化剂依达拉奉(edaravone,EDA)对低氧低糖处理后VSMC自噬和铁死亡发生以及细胞反应性的影响;同时采用创伤失血休克大鼠模型,观察抗氧化剂对休克血管功能的作用,以探讨抗氧化干预调节休克血管功能与低氧低糖VSMC反应性的作用,及其与细胞自噬和铁死亡的关系。
1 材料与方法
1.1 实验动物与试剂
清洁级SD大鼠62只,体质量180~220 g,由陆军特色医学中心实验动物中心提供,动物使用许可证号:SYXK(军)2017-0058。细胞培养用DMEM培养基和胎牛血清购自Gibco公司,EDA购自TOCRIS公司,兔源性微管相关蛋白1轻链3(microtubule associated protein 1 light chain 3,LC3)抗体和p62抗体购自Cell Signaling Technology公司,兔源性谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPx4)抗体和溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)抗体购自Abcam公司,小鼠源性β-actin抗体购自Invitrogen公司,山羊抗兔和抗小鼠荧光二抗购自Jackson公司,蛋白酶抑制剂片购自Thermo公司,荧光素标记的牛血清白蛋白(fluorescein isothiocyanate-conjugated bovine serum albumin,FITC-BSA)购自Sigma公司。其他试剂为国产分析纯。
1.2 原代大鼠VSMC培养及低氧低糖模型构建
在无菌条件下分离30只大鼠的肠系膜动脉,采用贴块法进行VSMC原代培养[13]。用含10%胎牛血清和1%双抗的完全培养基培养细胞至3~5代,用于后续实验。实验分为4组:正常对照组、低氧低糖组、低氧低糖+EDA 0.1 mmol/L组、低氧低糖+EDA 1 mmol/L组。正常对照组更换新鲜的正常培养基;低氧低糖组细胞用无血清的低糖DMEM培养基换液,置于低氧培养罐中培养12 h;低氧低糖+EDA组在缺氧前加入不同剂量的EDA。
1.3 Western blot检测细胞自噬标志物和铁死亡标志物表达水平
常规提取细胞蛋白并进行Western blot检测[13]。取各组样本用SDS-PAGE进行蛋白电泳,电泳条件为:4 ℃、恒定电压堆积胶80 V、分离胶100~120 V。然后转印至PVDF膜上,封闭后分别加入LC3抗体(1∶1 000)、p62抗体(1∶1 000)、GPx4抗体(1∶1 000)、SLC7A11抗体(1∶1 000)或β-actin抗体(1∶2 000)孵育。然后加入荧光标记二抗(1∶20 000)孵育,用双色红外成像激光系统(德国LI-COR公司)检测蛋白条带,用Quantity One分析软件测定条带灰度值。蛋白印迹检测重复3次。
1.4 细胞收缩反应性检测
将第3~5代的VSMC接种至Transwell小室,建立双室细胞培养模型[14]:Transwell小室插入24孔培养板,VSMC接种于上室,在上、下室加入完全培养基培养48 h后进行细胞收缩反应性检测。同上实验分组并处理后,在上室中加入FITC-BSA 5 mg/mL和去甲肾上腺素(norepinephrine,NE)10-7mmol/L,30 min后取下室液检测荧光强度,以FITC-BSA的渗透率变化反映VSMC的收缩反应性。
1.5 创伤失血休克大鼠模型构建及血管反应性检测
32只大鼠随机分为对照组、休克组、休克+EDA 100 μmol/L组、休克+EDA 200 μmol/L组,每组8只。采用本实验室常规方法建立大鼠创伤失血休克模型[15]:大鼠麻醉后给予右侧股骨骨折,然后左侧股动脉插管并连接水银血压计,放血使血压降至30 mmHg并维持2 h。模型完成后,开腹取肠系膜上动脉并制备血管环,采用离体血管张力测定技术检测血管反应性[13]。将平衡后的血管环用124 mmol/L高钾液诱导预收缩,记录预收缩张力(ΔK+),并将其作为量化标准。再次平衡后,休克+EDA组加入不同剂量的EDA孵育30 min,然后加入10-9~10-5mmol/L梯度浓度的NE,记录NE诱导血管环产生的收缩张力(ΔNE),用量—效曲线和标准化的最大收缩张力(Emax=ΔNE/ΔK+)评价血管反应性。
1.6 统计学方法

2 结果
2.1 EDA对低氧低糖处理后VSMC自噬水平的影响
低氧低糖处理12 h后,VSMC中自噬标志物LC3-Ⅱ/LC3-Ⅰ比值升高、p62蛋白表达水平降低,与正常对照组比较,差异有显著统计学意义(P<0.01)。EDA处理可使低氧低糖细胞的LC3-Ⅱ/LC3-Ⅰ比值降低、p62蛋白表达水平升高,与低氧低糖组比较,差异有统计学意义(P<0.05) ,其中1 mmol/L的EDA效果更为显著(P<0.01),见图1。
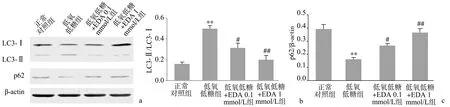
a:代表性免疫印迹条带;b:LC3-Ⅱ/LC3-Ⅰ比值;c:p62/β-actin比值 **:与正常对照组比较,P<0.01;#:与低氧低糖组比较,P<0.05;##:与低氧低糖组比较,P<0.01
2.2 EDA对低氧低糖处理后VSMC铁死亡发生的影响
低氧低糖处理后,VSMC中铁死亡标志蛋白GPx4和SLC7A11的表达水平降低,与正常对照组比较,差异有统计学意义(P<0.05),提示铁死亡发生。而EDA处理后低氧低糖状态下GPx4和SLC7A11的表达水平与低氧低糖组比较,差异无统计学意义(P>0.05),见图2。
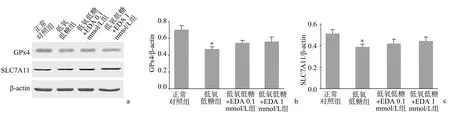
a:代表性免疫印迹条带;b:GPx4/β-actin比值;c:SLC7A11/β-actin比值 *:与正常对照组比较,P<0.05
2.3 EDA对低氧低糖处理后VSMC收缩反应性的影响
低氧低糖处理后VSMC对NE诱导的收缩反应性明显降低,荧光标记物渗透率显著低于正常对照组(P<0.01)。EDA处理可使低氧低糖VSMC的收缩反应性明显升高,低氧低糖+EDA 0.1 mmol/L组和低氧低糖+EDA 1 mmol/L组的荧光渗透率均高于低氧低糖组(P<0.05),见图3。

**:与正常对照组比较,P<0.01;#:与低氧低糖组比较,P<0.05;##:与低氧低糖组比较,P<0.01
2.4 EDA对创伤失血休克大鼠血管反应性的影响
创伤失血休克使大鼠血管对NE诱导的收缩反应性明显降低(P<0.01)。给予EDA处理后,休克血管的收缩反应性明显升高,使血管对NE的量—效曲线左移、Emax升高,与休克组相比差异有统计学意义(P<0.05),见图4。

a:量—效曲线;b:Emax **:与对照组比较,P<0.01;#:与休克组比较,P<0.05;##:与休克组比较,P<0.01
3 讨论
细胞自噬和铁死亡是不同于经典细胞凋亡和坏死的新的细胞死亡途径,它们在形态特征、生化特点和基因调控等方面都有着明显不同[16]。细胞自噬作为广泛存在于真核细胞内的一种基本生命现象,与多种疾病(如癌症、神经系统疾病、代谢性疾病等)密切相关[3-6]。因此,通过调控自噬来探索疾病治疗的新方法成为了当前研究的重点问题。然而自噬与多种致病因素间的相互作用和调节机制及其与疾病发生发展的关系目前尚未明确。对铁死亡的研究更是处于初期阶段,尽管铁死亡与多种临床疾病间的关系都得到了证实,但其具体作用和调节机制仍不清楚,其与血管功能调节和疾病状态下血管功能障碍的相关性也少见报道。
氧化应激是指机体内氧化和抗氧化作用失衡而导致氧化损伤的过程。目前已经证实,氧化应激是多种疾病中器官系统损伤的重要病理机制之一。近年来发现,氧化应激与自噬之间有紧密的联系,如对动脉粥样硬化、缺血性心脑疾病的研究发现,氧化应激作为上游刺激因素可诱导保护性的自噬反应,自噬激活后通过清除损伤的蛋白和细胞器降低氧化损伤,从而发挥细胞保护作用[17]。而Hariharan等[18]报道,缺血/再灌注损伤引起了小鼠心肌氧化应激和自噬,抗氧化剂治疗能抑制氧化应激和自噬并减少心肌梗死面积,因此其认为氧化应激介导的过度自噬激活是心肌缺血/再灌注损伤的重要因素。这些氧化应激与自噬相互作用及其对细胞不同影响的研究结果提示,不同的应激水平和强度与不同的疾病病理生理阶段可能导致不同的自噬反应和细胞结局。铁死亡发生的主要机制是铁依赖性的脂质氧化应激,其生化特征即表现为铁和自由基聚集、谷胱甘肽耗竭、GPx4活性降低、脂质过氧化损伤发生[8-10]。本实验室前期研究显示,严重创伤失血休克后,缺血缺氧损伤导致的氧化应激反应在创伤后器官功能障碍中起重要作用[13]。但是,创伤失血休克引起的氧化应激反应是否会激活自噬和铁死亡、参与血管功能障碍的发生,调节氧化反应能否通过影响细胞自噬或铁死亡来发挥作用,目前尚不清楚。
本实验通过对原代培养的VSMC给予低氧低糖处理,模拟在体创伤失血休克后缺血缺氧状态,观察抗氧化干预对低氧低糖诱导VSMC自噬和铁死亡以及细胞收缩功能的影响,结果显示,低氧低糖处理12 h后,VSMC的自噬水平升高、铁死亡发生、细胞收缩反应性降低;抗氧化剂EDA处理可抑制自噬水平的升高、增加细胞收缩反应性,但对铁死亡标志蛋白水平无明显影响。随后本研究采用休克动物模型作功能验证,结果显示抗氧化剂EDA处理可改善休克引起的血管收缩功能下降。提示抗氧化干预可抑制低氧低糖诱导的VSMC自噬激活,并对缺氧细胞和休克血管的收缩功能有改善作用。但本实验中未发现抗氧化剂EDA处理对铁死亡相关蛋白水平有明显作用,其具体原因尚需进一步研究。尽管氧化应激在多种心血管疾病病理生理过程中的重要作用已得到广泛认识,抗氧化治疗也备受关注,但目前抗氧化剂在心血管疾病治疗中的效果并不符合预期。因此,仍需对机体氧化损伤的机制作进一步的探索,如氧化反应与自噬间的相互作用机制、参与疾病病理过程的关键靶点等,以期为抗氧化措施在临床疾病治疗中的应用、更有效的抗氧化剂的开发提供研究基础。
