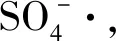Ti4O7多孔膜电极降解橙黄Ⅱ的效果与催化机制
2022-01-24张云澍高庆伟赵庆良
张云澍,丁 晶,高庆伟,王 琨,赵庆良
(1.上海理工大学 环境与建筑学院,上海 200093;2.哈尔滨工业大学 环境学院,哈尔滨 150090)
随着中国工业化的迅速发展,染料废水的排放呈逐年增加趋势,若随意排放则会对环境造成严重的污染[1]。橙黄Ⅱ是一种典型的偶氮染料,其废水具有色度高、毒性强、难降解等特点,用传统的生物法难以有效处理[2]。目前,染料废水常用的处理方法包括膜过滤法、氧化法和吸附法等,其中EAOPs(electrochemical advanced oxidation processes,电化学高级氧化)通过电极表面发生的氧化还原反应,直接或间接地氧化降解水体中的有机污染物,具有降解效率高、二次污染少、可控性强等优势,被广泛用于染料废水的处理[3-4]。
Ti4O7电极具有导电性强、耐腐蚀性强、析氧电位高和成本相对低廉的特点,常用于电催化降解水中的PPCPs(pharmaceutical and personal care products,药物及个人护理品)、全氟化合物、偶氮染料等有机污染物,是目前最具有应用前景的电极材料[5]。然而,目前Ti4O7电极的制作通常采用真空热压或等离子喷涂工艺,成本极高[6-8]。此外,污染物向电极表面的传质过程是限制污染物电催化降解的重要因素[9]。近年来,研究发现电解液穿流通过电极可以加速污染物向电极表面的传质,进而加速污染物的降解并增加电流效率[10-12]。
为了降低Ti4O7电极的制作成本并增强有机污染物向Ti4O7电极表面的传质,拟采用提拉浸渍-氢热还原法制备非对称Ti4O7多孔膜电极,并以Ti4O7多孔膜电极为基础构建穿流-电催化氧化系统,进而分析橙黄Ⅱ在穿流-电催化氧化作用下的降解动力学,研究Ti4O7多孔膜电极的稳定性并揭示Ti4O7多孔膜电极的催化机制。
1 实 验
1.1 Ti4O7多孔膜电极的制备方法
TiO2溶胶的配制如下(g/100 mL):PVA,10;TiO2,20;甘油,1.5;N-甲基吡咯烷酮,1;聚丙烯酸,2。采用提拉浸渍法在陶瓷微滤膜(50 mm×30 mm×5 mm)基底上涂覆一层TiO2溶胶膜,随后将TiO2溶胶膜置于常温常湿条件下干燥48 h形成TiO2凝胶膜,而后在800 ℃条件下高温退火4 h,取出并冷却至室温即得到TiO2陶瓷膜。将TiO2陶瓷膜至于管式炉中,在H2(120 mL/min)气氛下900 ℃退火4 h,冷却后即制成Ti4O7多孔膜电极。
1.2 实验装置和运行
采用偶氮染料橙黄Ⅱ作为特征性有机污染物,并配制成橙黄Ⅱ溶液(0~50 mg/L,0.1 mol/L Na2SO4,pH为7)用于电解实验。非穿流电解实验在烧杯(250 mL)中进行,Ti4O7多孔膜电极作为阳极,钛片(50 mm×30 mm)作为阴极,两极间距20 mm,采用磁力搅拌器(800 r/min)加速电解过程中橙黄Ⅱ向电极表面的传质。穿流-电催化氧化装置如图1所示,实验装置均由有机玻璃制成,进水经过隔膜泵提高水压,采用三通阀门调节管道压力、管道压力表监测管道压力。电解过程中橙黄Ⅱ溶液通过循环流的方式进入电解系统,Ti4O7多孔膜电极作为阳极,钛片(50 mm×30 mm)作为阴极,两极间距20 mm。电解过程所需的电流(2~10 mA/cm2)由直流稳压电源(RPS3003D-2)提供。
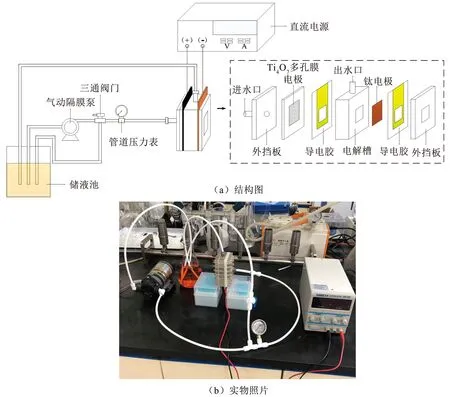
图1 穿流-电催化氧化系统的结构与实物照片Fig.1 Structure diagram and photo of flow-through electrochemical system
1.3 分析方法
采用SEM(scanning electron microscope,扫描电子显微镜,Mira3)对Ti4O7多孔膜电极的表面和截面进行显微观察。采用XRD(X-rays diffraction,X-射线衍射,Ultima IV)分析膜电极表面的晶体组成。采用氮吸附仪(Autosorb iQ)测定Ti4O7多孔膜电极的N2吸附-解吸等温线,并通过多点BET(Brunauer-Emmett-Teller)法计算Ti4O7多孔膜电极的比表面积。采用压汞法(Micromeritics AutoPore IV)测定Ti4O7多孔膜电极的孔隙率和孔径。通过测试Ti4O7多孔膜电极在Na2SO4溶液(0.1 mol/L,pH为7)中的极化曲线,计算Ti4O7多孔膜电极的析氧电位。测试条件是:以Ti4O7陶瓷片电极为工作电极(2 cm×2 cm),铂片(1.5 cm×1.5 cm)和Hg/Hg2SO4分别作为对电极和参比电极,扫描电位为1~3 V (vs.SHE),扫描频率为1 mV。采用电子顺磁共振波谱仪(EPR)监测Ti4O7多孔膜电极在Na2SO4溶液(100 mmol/L,pH为7)电解过程中产生的自由基,并采用DMPO(5,5-Dimethyl-1-pyrrolineN-oxide)作为自由基捕获剂。采用紫外分光光度计(UV-2450)测定溶液中橙黄Ⅱ的特征吸收峰和质量浓度。采用总有机碳分析仪(VSCN8)测定溶液中的溶解性有机碳(dissolved organic carbon,DOC)。
1.4 拟合与计算
橙黄Ⅱ在电解过程中的降解动力学采用准一级动力学模型进行拟合,准一级动力学的公式为
(1)
式中:ρt为不同电解时间条件下溶液中橙黄Ⅱ的质量浓度,mg/L;ρ0为溶液中橙黄Ⅱ的初始质量浓度,mg/L;k1为准一级动力学速率常数,min-1;t为电解反应时间,min。
电流矿化效率(mineralization current efficiency,EMC)的计算公式为
(2)
式中:EMC为电解过程的电流矿化效率,%;n为有机化合物完全矿化过程的理论电子转移数;F为法拉第常数,F=96 485 C/mol;ΔDOC为电解过程中溶液DOC的降解量,mg;Vs为电解溶液的体积,L;4.32×107为单位均化的转换因子,s·mg/(h·mol);m为橙黄Ⅱ中的碳原子数;I为施加电流,A。
单位质量DOC降解能耗Cm,DOC的计算公式为
(3)
式中Ecell为电解过程中平均电压,V。
2 结果与讨论
2.1 Ti4O7多孔膜电极的表征
TiO2晶体在H2气氛下热还原的过程中,晶体内部的氧空位逐渐增加,逐渐从金红石型转变为Magnéli相(TinO2n-1,4≤n≤10)。如图2所示,在退火温度为900 ℃、退火时间为4 h的条件下,TiO2被H2还原,每隔4层金红石结构单元产生一个共用氧原子的剪切面,形成氧空位,转变成高纯相的Ti4O7晶体。氧空位的产生改变了原有晶体的核外电子轨道,使Ti4O7具有导体的性质。此外,氧空位可以产生晶格缺陷,提供催化位点。
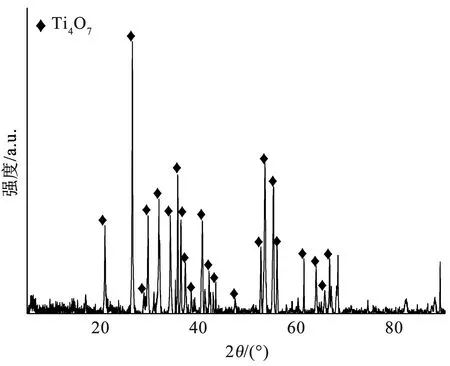
图2 Ti4O7多孔膜电极的XRD图谱Fig.2 XRD patterns of Ti4O7 porous membrane electrode
通过SEM观察Ti4O7多孔膜电极表面和截面的微观结构,结果如图3所示。陶瓷微滤膜基底与Ti4O7膜的微观结构差异非常明显,陶瓷微滤膜基底是由不规则陶瓷颗粒堆积而成,Ti4O7膜则是由直径约1 μm的规则Ti4O7颗粒热烧结形成的(图3(a))。Ti4O7膜层紧密烧结在陶瓷微滤膜表面,膜层分布均匀,厚度为48~51 μm(图3(b))。Ti4O7膜层表面粗糙,Ti4O7颗粒在退火过程中部分烧结在一起,形成均匀分布的孔状结构(图3(c))。

图3 Ti4O7多孔膜电极的SEM呈像Fig.3 SEM images of Ti4O7 porous membrane electrode
通过N2吸脱附实验分析了Ti4O7多孔膜电极的比表面积和孔隙特征,结果如图4所示。Ti4O7多孔膜电极的N2吸脱附等温线与其他大孔无机非金属氧化物材料相似,符合Ⅲ型等温线(图4(a))[13]。Ti4O7多孔膜电极在低压区对N2的吸附速率较低,而在高压区对N2的吸附速率明显提高。此外,等温曲线没有明显的滞回环,证明了Ti4O7多孔膜电极的介孔分布极少。Ti4O7多孔膜电极的比表面积为10.18 m2/g,相比平板电极,多孔电极具有更高的比表面积,可以提供更多的催化位点,增强电极的电催化性能。Ti4O7多孔膜电极的孔径分布集中,主要分布在0.1~1.0 μm(图4(b)),符合微滤膜的孔径标准[14]。此外,Ti4O7多孔膜电极的孔隙率为47.63%,具有较高的膜通量。
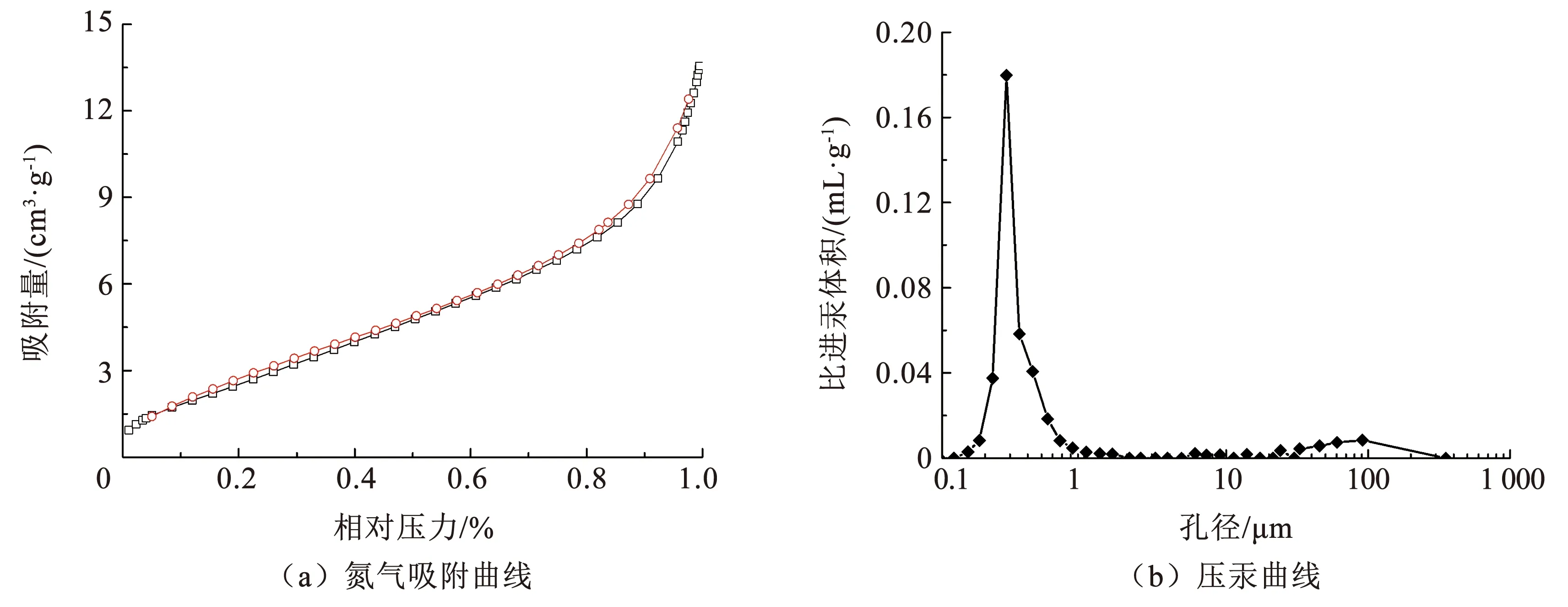
图4 Ti4O7多孔膜电极的氮气吸附曲线和压汞曲线Fig.4 Nitrogen adsorption curves and mercury pressure curves of Ti4O7 porous membrane electrode
已有研究将多孔Ti4O7陶瓷电极用于电催化降解水中的全氟辛烷磺酸,电极的孔隙率为21.6%,孔径为2.8~3.6 μm,全氟辛酸在Ti4O7陶瓷电极作用下的降解率高达93.1%,远高于Ce掺杂的PbO2电极和BDD电极[15]。本研究制备的Ti4O7多孔膜电极的孔径分布更窄,孔隙率更大,因此,推断其具有较强的电催化效果。
通过线性伏安扫描获得了Ti4O7多孔膜电极的极化曲线,如图5所示。Ti4O7多孔膜电极在pH为中性的硫酸钠溶液(0.1 mol/L)中的析氧电位可达2.2 V(vs.SHE)。表1为不同电极的析氧电位,以Pt电极作为标准,析氧电位低于Pt电极的称为活性电极,而析氧电位高于Pt电极的称为非活性电极。活性阳极表面分子的轨道中存在大量孤对电子,容易被电极表面吸附的·OH(羟基自由基)氧化,最终形成氧气;而非活性阳极表面分子轨道中的电子在各轨道中分布稳定,·OH通常以物理吸附的方式吸附在电极表面。·OH的标准氧化还原电位高达2.8 V(vs.SHE),是EAOPs中最重要的氧化剂[16]。因此,Ti4O7多孔膜电极的析氧电位与BDD电极相近,是一种非活性电极,适用于EAOPs的阳极[17]。
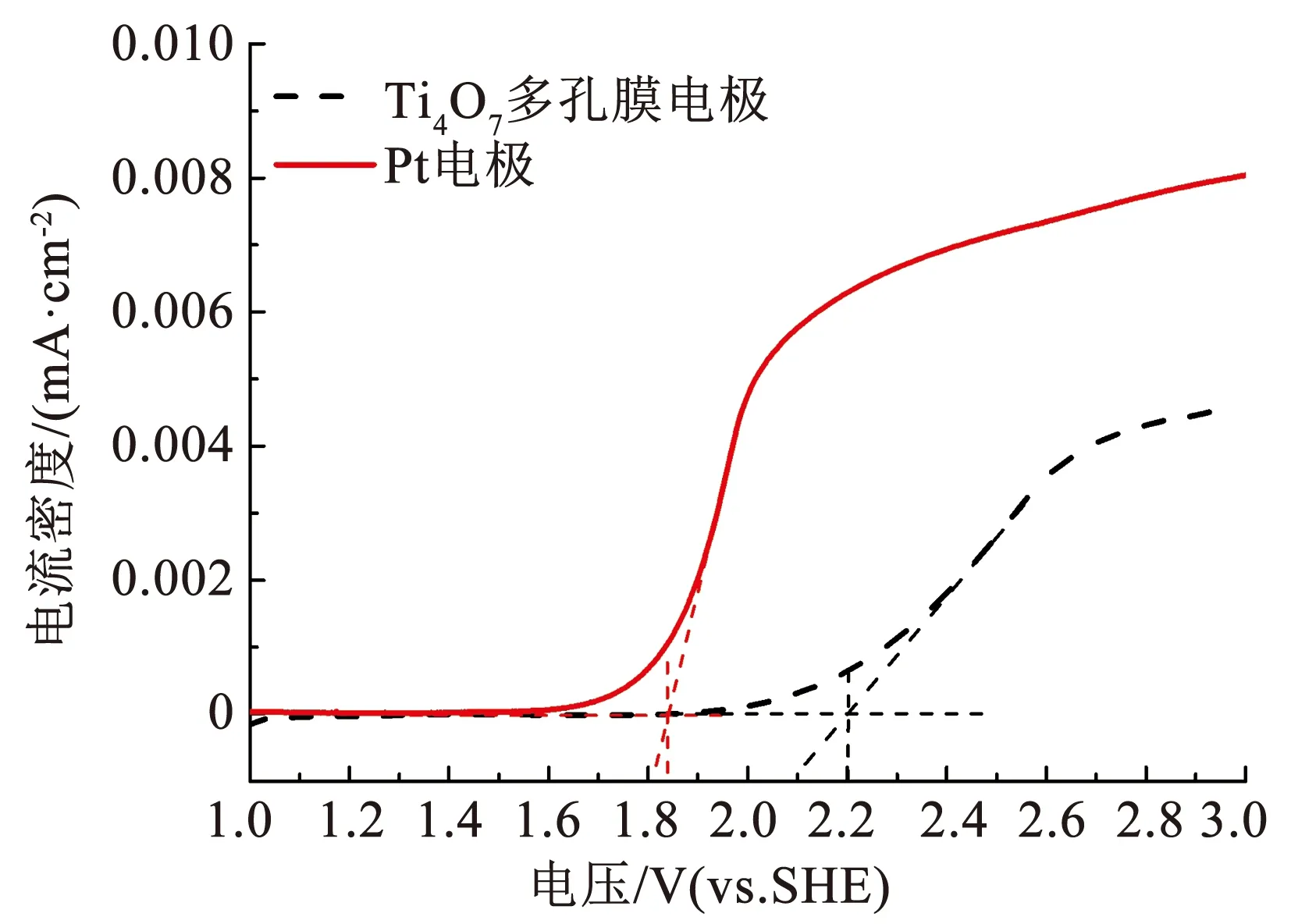
图5 Ti4O7多孔膜电极的极化曲线Fig.5 Polarization curves of Ti4O7 porous membrane electrode

表1 不同电极的析氧电位Tab.1 Oxygen evolution potential of different electrodes
2.2 Ti4O7多孔膜电极对橙黄Ⅱ的降解效果分析
2.2.1 穿流和非穿流模式下橙黄Ⅱ的降解效果对比
在电流密度为5 mA/cm2、极间距为2 cm的条件下,采用穿流和非穿流模式电催化降解橙黄Ⅱ,结果如图6所示。相比非穿流模式,橙黄Ⅱ溶液在穿流模式下的降解过程中脱色效果更为显著,非穿流模式下溶液中橙黄Ⅱ的终质量浓度降低至(15.08±0.63)mg/L,降解率为69.85%,而在穿流模式下溶液中橙黄Ⅱ的终质量浓度降低至(4.49±0.82)mg/L,降解率为91.03%(图6(a))。穿流和非穿流模式下,橙黄Ⅱ降解曲线的准一级动力学拟合相关系数(R2)分别为0.97和0.99(图6(b)),说明穿流模式和非穿流模式下溶液中橙黄Ⅱ的降解过程符合准一级动力学。穿流模式和非穿流模式下橙黄Ⅱ降解过程的k1分别为0.055 4和0.023 3 min-1,溶液中橙黄Ⅱ半衰期(t1/2)分别为12.51和29.75 min。穿流和非穿流模式下,在60 min的电解过程中溶液中DOC的终质量浓度分别为6.74和12.58 mg/L,DOC降解率分别为74.23%和51.61%(图6(c))。穿流和非穿流模式下溶液中橙黄Ⅱ的EMC分别为88.77%和62.08%,溶液中橙黄Ⅱ的Cm,DOC分别为0.49和0.66 kWh/g。
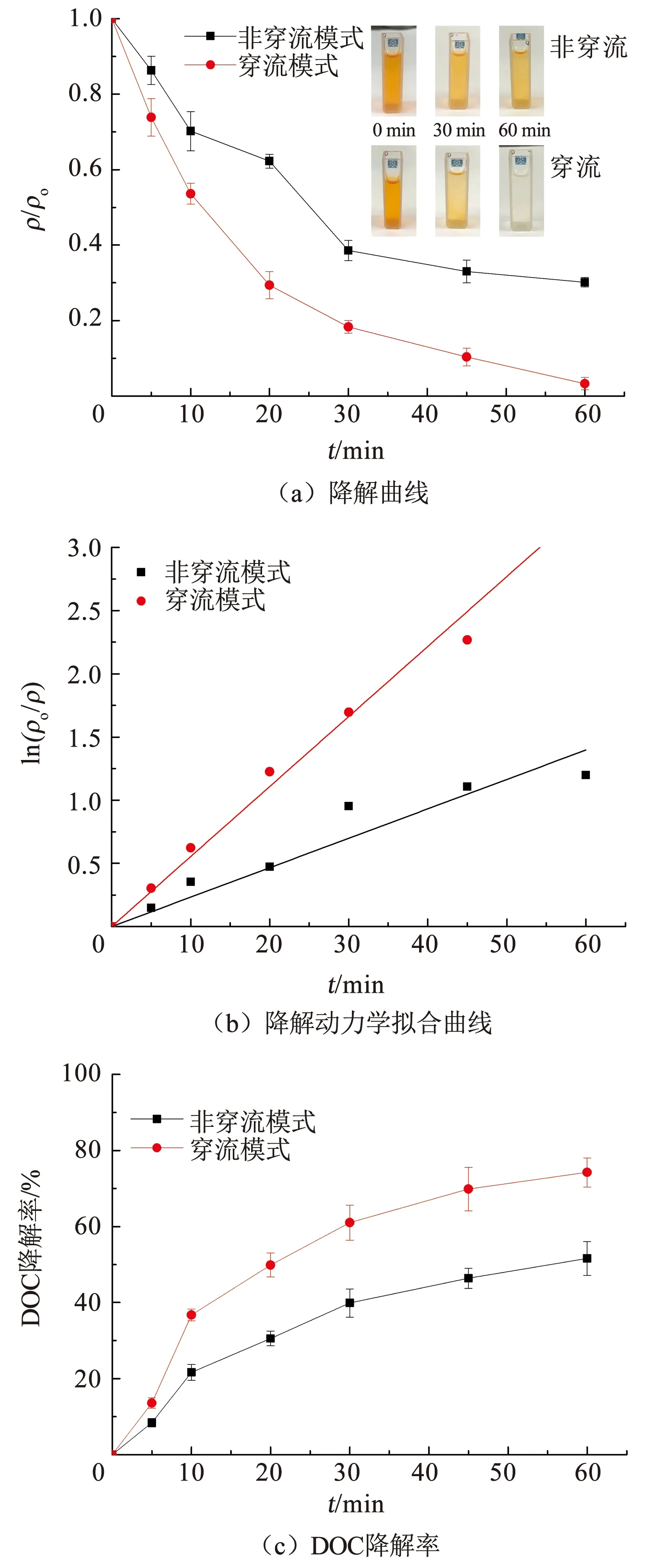
图6 橙黄Ⅱ在穿流和非穿流模式下的降解曲线和降解动力学拟合曲线Fig.6 Fitting of orange Ⅱ degradation curves and kinetics curves in flow-through and non-flow-through modes
橙黄Ⅱ的电催化降解过程主要包括橙黄Ⅱ向电极表面的传质和橙黄Ⅱ在电极表面的电子转移。橙黄Ⅱ的电子转移过程主要是与电极表面产生的·OH之间发生氧化还原反应,进而被氧化降解。然而,·OH在水中的存活时间仅有几纳秒,因此,污染物向电极表面的传质过程通常是污染物的电催化降解的限速过程[18]。非穿流模式和穿流模式下的电流密度和两极间距是相同的,说明两种模式下污染物在电极表面的电子转移过程基本一致。而在穿流模式下,溶液中橙黄Ⅱ的降解速率远高于非穿流模式,证明了穿流模式具有更高效的污染物液相传质效率。其他研究比较了非穿流和穿流模式下层状Ti4O7电极对苯达松的电催化降解效果,发现在非穿流模式下,6 h后苯达松的降解率仅为7%,而在穿流模式下,6 h后苯达松的降解率高达85%[19],说明穿流模式可以增强苯达松与电极表面间的传质,与本研究观点一致。
2.2.2 穿流-电催化氧化系统的反应条件对橙黄Ⅱ降解的影响
为了考察不同的反应条件穿流-电催化氧化系统对橙黄Ⅱ降解的影响,分别分析了不同管道压力、电流密度、橙黄Ⅱ初始质量浓度和pH条件下橙黄Ⅱ的降解效果,结果如图7和表2所示。

表2 橙黄Ⅱ在不同反应条件下降解的准一级动力学拟合参数Tab.2 Pseudo first-order kinetic fitting parameters for orange II degradation
随着管道压力从0.02 MPa升高至0.10 MPa,Ti4O7多孔膜电极的膜通量从0.012 cm/s升高至0.021 cm/s,反映了橙黄Ⅱ向Ti4O7多孔膜电极表面的传质速率增加。受传质的影响,溶液中橙黄Ⅱ的降解速率也随管道压力的提高而增加,k1从0.044 min-1增加至0.060 min-1,橙黄Ⅱ的终质量浓度从3.55 mg/L降低至1.40 mg/L(图7(a))。
电流密度对橙黄Ⅱ降解的影响非常显著,随着电流密度从2 mA/cm2升高至10 mA/cm2,溶液中橙黄Ⅱ的终质量浓度从12.3 mg/L降低至1.50 mg/L;橙黄Ⅱ降解过程的k1从0.023 min-1增加至0.057 min-1,提高了1.48倍(图7(b))。已有研究采用热压法制备了多孔Ti4O7电极并催化降解四环素溶液,发现当系统的电流密度提升至2 mA/cm2后,溶液中四环素的降解速率不再随着电流密度的提升而增加[20]。在传统的电催化系统中,随着电流密度的增加,电极表面的电子转移逐渐增强,产生更多的·OH和空穴等氧化物加速污染物的降解。然而,随着电极表面污染物的快速消耗,污染物向电极表面的传质过程逐渐代替污染物在电极表面的电子转移过程,成为污染物降解的限速步骤。Ti4O7多孔膜电极在穿流模式下可以极大地增强污染物向电极表面的传质。因此,在穿流模式下,随着电流密度的提升,溶液中橙黄Ⅱ的降解速率可以显著地增加。
当橙黄Ⅱ的质量浓度从10 mg/L增加至20 mg/L时,橙黄Ⅱ降解过程的k1从0.069 min-1降低至0.057 min-1。而随着初始橙黄Ⅱ质量浓度的继续增加,橙黄Ⅱ降解过程的k1变化很小(图7(c))。在传统的电催化氧化系统中,随着污染物初始质量浓度的持续升高,污染物降解的准一级动力学速率常数显著下降,反映了污染物向电极表面的传质是污染物电催化降解的限速步骤[21]。Ti4O7多孔膜电极具有较高的比表面积,在电催化过程中可以提供更多的催化位点,降低电子转移过程对污染物电催化降解的限制。此外,穿流模式下可以极大程度地增强橙黄Ⅱ向电极表面的传质,降低了传质过程对污染物电催化降解的限制。因此,高质量浓度橙黄Ⅱ溶液降解过程的k1基本不变,说明橙黄Ⅱ的降解没有受到电子转移过程和传质过程的限制。
溶液的pH对橙黄Ⅱ电催化降解过程的影响较小,经过60 min的电催化降解后,不同pH溶液中橙黄Ⅱ的降解率均在95%以上。随着溶液中pH的增加,橙黄Ⅱ降解过程的k1从0.060 min-1缓慢降低至0.050 min-1(图7(d))。在阳极氧化过程中,析氧反应是重要的副反应,如式(4)所示:
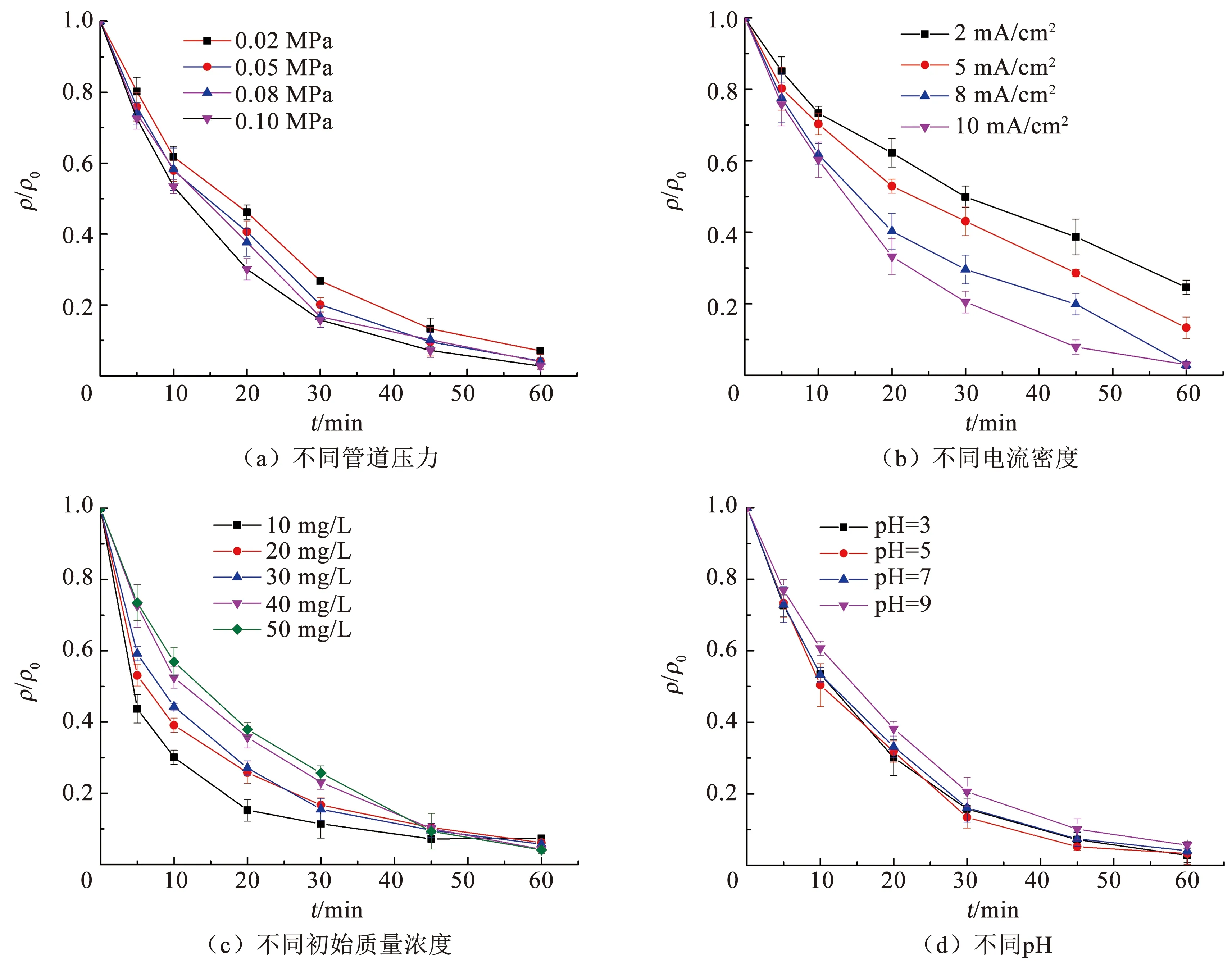
图7 橙黄Ⅱ在不同反应条件下的降解曲线Fig.7 Orange II degradation curves in different conditions

(4)
在低pH条件下,溶液中OH-的活度偏低,阳极的析氧过电位升高,析氧反应受到抑制,电催化产生·OH的反应效率升高,污染物的降解速率增加。而随着pH的增加,溶液中OH-的活度逐渐升高,阳极的析氧过电位逐渐降低,析氧反应逐渐活跃,抑制了电催化产生·OH的反应,污染物的降解速率降低。此外,阳极的析氧反应会在阳极表面形成大量的微气泡,影响污染物向阳极表面的传质,也会导致污染物电催化降解速率的降低。
2.3 Ti4O7多孔膜电极的稳定性和催化机制分析
2.3.1 Ti4O7多孔膜电极的稳定性分析
通过循环降解实验验证Ti4O7多孔膜电极的稳定性,结果如图8所示。相比初始Ti4O7多孔膜电极的表面形貌,循环10次后的膜电极表面会产生大量坑洼,这是由穿流降解过程和超声清洗过程产生的水力冲击所致。循环前后Ti4O7多孔膜电极表面的物相变化(图9)表明,相比初始Ti4O7多孔膜电极,循环10次后膜电极表面的物相组成仍为纯相Ti4O7晶体,但Ti4O7晶体的结晶度相比初始的膜电极略低,这是由于水力冲击导致Ti4O7多孔膜电极表面损伤,使得Ti4O7晶体部分流失并露出底层的非晶陶瓷膜,在XRD衍射结果中体现为较低的结晶度。10次循环降解过程中橙黄Ⅱ的降解效率基本一致,说明膜电极在多次循环下具有良好的稳定性(图10),虽然在水力冲击的条件下会导致表面Ti4O7晶体的损失,但电极表面仍存在足够的催化位点,橙黄Ⅱ降解过程不会受电子转移步骤控制。

图8 原始与循环10次后Ti4O7多孔膜电极的表面形貌Fig.8 Surface images of Ti4O7 porous membrane electrode before and after 10 cycles
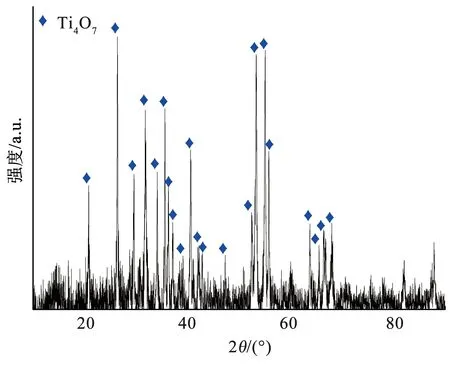
图9 循环10次后Ti4O7多孔膜电极的XRD图谱Fig.9 XRD patterns of Ti4O7 porous membrane electrode after 10 cycles
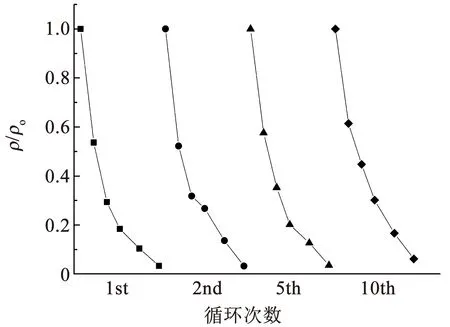
图10 不同循环次数下橙黄Ⅱ的降解曲线Fig.10 Orange II degradation curves under different cycles
2.3.2 Ti4O7多孔膜电极的催化机制分析
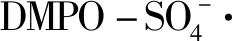
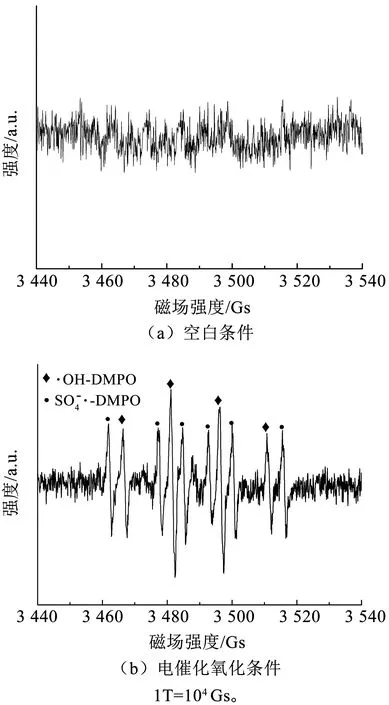
图11 空白和电催化氧化过程电极表面的EPR图谱Fig.11 EPR spectrum of electrode surface of blank and electrochemical oxidation process
(5)
(6)
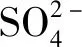
3 结 论
1)采用溶胶凝胶-氢气热还原法制备了Ti4O7多孔膜电极,其具有较高的比表面积、集中的孔径分布、较高的孔隙率,是一种功能性的陶瓷微滤膜;Ti4O7多孔膜电极的析氧电位高达2.2 V(vs.SHE),属于非活性电极。
2)穿流-电催化氧化系统可以显著增强橙黄Ⅱ向Ti4O7多孔膜电极表面的液相传质并加速橙黄Ⅱ的降解,橙黄Ⅱ在非穿流和穿流模式下的降解过程均符合拟一级动力学,反应速率常数分别为0.023和0.055 min-1,电流效率分别为62.08%和88.77%。