16S rRNA基因可变区与全长序列进化关系相似性分析
2022-01-23刘爽爽刘峰辉
刘爽爽, 帖 云, 齐 林, 刘峰辉, 王 磊
(1. 郑州大学 信息工程学院 河南 郑州 450001; 2. 郑州大学第一附属医院 河南 郑州 450052; 3. 河南省人民医院 口腔医学中心 河南 郑州 450003)
0 引言
16S rRNA基因是微生物生态学分析中最常使用的一类分子标志物,存在于所有细菌的基因组中,具有高度的特异性和保守性,序列长度约为1 500 bp[1]。除保守区外,16S rRNA基因序列还存在V1~V9共9个可变区[2],不同可变区的长度范围为100~300 bp,新一代测序技术可以使用短配对碱基轻易覆盖,使得 16S rRNA 序列可变区的测量更加便捷。
可变区的特异性能够反映出不同微生物的特征核苷酸序列,用于分析复杂生物环境中微生物的物种多样性[3]、相对丰度[4]、物种鉴定及进化距离等[5]。文献[6]对16S rRNA 基因两个可变区进行了比较研究,结果显示,在肠道菌群物种多样性分析及物种鉴定能力上,选择V1~V3可变区片段进行测序,得到了与全长序列更为接近的结果。文献[7]利用16S rRNA全长序列和部分基因作为热测序的靶点,在不同水平上分析了16S rRNA基因在基因组内因异质性引起的高估问题,结果表明,对于细菌使用针对V4和V5可变区的引物可以将这种高估最小化。文献[8]采用Illumina Miseq测序技术,测定了苏尼特和阿拉善双峰驼的自然发酵驼乳中微生物16S rRNA的V3、V4可变区序列,并对群落结构和物种多样性进行了比较分析。文献[9]对HIV-1包膜蛋白gp120进行分析时,找到了可变区V1可能作为传播选择靶位点的证据。文献[10]分析了大西洋鲑鱼细菌16s rRNA基因全长序列及不同可变区对微生物群落结构的影响,发现不同可变区对微生物分布和系统发育有着不同的影响。目前可变区在物种进化关系中表现如何的研究较少,但其对物种进化来源分析具有重要的指导意义。本文以核糖体数据库项目(RDP)所提供的细菌16S rRNA基因数据为基础,构建不同可变区及全长序列进化树,使用层次距离矩阵算法分析了V2、V3、V4可变区与全长序列所构建的进化树之间的距离差异值,并对可变区与全长序列进化关系的相似性进行了分析。
1 数据获取与预处理
1.1 可变区截取与筛选
原始数据采用RDP中细菌16S rRNA的全部序列。压缩文件大小为3 GB,解压后大小为76 GB,共包含约320万条16S rRNA序列,数据格式为fasta。由于V9区在实际研究中应用较少,故只选用V1~V8可变区进行相关研究。使用MEGA6软件在该序列中分别寻找各可变区两侧保守序列,保守序列及可变区位置如表1所示。

表1 可变区两侧保守序列及位置
确定了可变区位置后,使用biopython函数库(https:∥biopython.org/)中的序列切片方法进行可变区片段的截取。初步截取后的序列中仍存在一些含有实际碱基数目较少且信息量较低的序列,因此需要分析可变区片段实际碱基长度并筛选出含有一定信息量的序列。使用biopython库中的seq.parse函数读取初步处理后的序列,统计每个序列中的实际碱基数目,并使用matplotlib库(https:∥matplotlib.org/)绘制可变区各序列的实际碱基数目分析图,以便对序列进行初步筛选。对序列的初步处理操作由python脚本完成,所使用的核心函数库为biopython库。
8个可变区使用相同的方法进行实际碱基数目统计,以V2可变区为例,实际碱基数目结果如图1所示。可以看出,可变区片段中实际碱基数目出现了明显的拐点,有约70万个V2可变区片段中碱基缺失较为严重。将拐点处放大,可以观察到部分V2可变区片段实际碱基数目在80以下,表明在这些序列的测序过程中,V2可变区的测序出现了遗漏或者并未对V2可变区进行测序。因此,需要按照拐点处实际碱基数目对约300万个可变区片段进行筛选,以保留含有一定信息量的可变区片段。
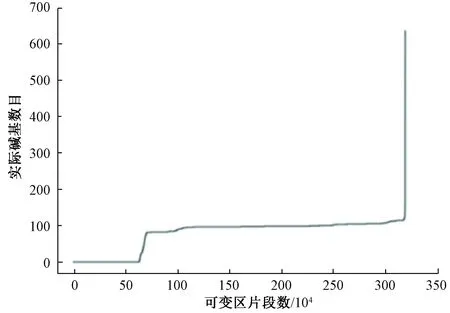
图1 V2可变区实际碱基数目
筛选操作仍由python脚本完成,在完成了8个可变区片段的初步提取和筛选后,就得到了可使用命令行工具进行处理的数据。对提取出的可变区片段进行去冗余与去除嵌合体操作,以V2可变区为例,两端对齐可以发现,序列数目为2 487 500,片段长度为1 264 bp,实际碱基数目为80~120,分别使用unique.seqs与chimera.uchime函数去除冗余部分序列和包含嵌合体较多的序列,再次对序列进行总结分析,此时序列数目缩减为533 136,片段长度缩减为771 bp。在完成了所有可变区的筛选、过滤操作后,绘制8个可变区预处理前后序列碱基数目对比图,结果如图2所示。通过可变区截取、筛选等数据预处理后,从序列碱基数目对比图可以看出,V2、V3、V4可变区预处理后序列碱基数目较其他可变区多,且V2、V4可变区序列数目比V3可变区多,V2、V4两个可变区较其他可变区包含更多的序列信息。
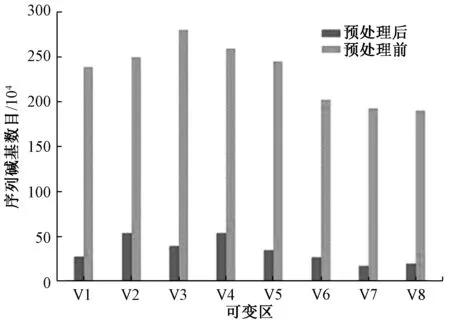
图2 可变区预处理前后序列碱基数目对比
1.2 可变区OTU聚类
为了方便区分序列,不同的16S rRNA基因序列若相似性高于97%,就可以把它定义为一个操作分类单元(operational taxonomic unit,OTU),每个OTU对应于一个不同的微生物种。通过OTU聚类分析可以简化数据结构,得到样品中微生物多样性水平以及不同微生物的丰度。对各可变区分别按相似度97%、98%及100%进行OTU聚类,取均值作为各可变区OTU数目,对比结果如图3所示。通过可变区聚类后的OTU数目可以看出,V2、V4两个可变区在多样性水平上较其他可变区更接近全长序列。

图3 可变区聚类后OTU数目对比
通过以上分析可以看出,V2、V4可变区在含有独特序列数目和实际碱基长度上均优于其他6个可变区,使用包含V2、V4可变区的基因测序片段对序列进行分类,将得到更加接近全长序列的分类结果。
2 进化关系相似性分析
2.1 进化树的构建
在各个可变区分析结果的基础上,为了更好地反映可变区对物种进化关系的相似性且尽可能减少计算的复杂度,选用的数据必须在门、纲、目、科、属、种层次上具有良好的区分度。使用RDP网站的Browser在数据库中选取了56条序列,这些序列来自11种不同门、16种不同纲的56个不同属种的细菌。为了将V2、V4可变区与其他可变区进行对比分析,基于OTU聚类的结果,选取V3可变区作为对照组,分别对V2、V3、V4可变区构建进化树,并与全长序列进化树进行比较。为了准确截取出V2、V3、V4可变区,将一条细菌序列添加到多重比对中,使用MEGA软件搜索特定保守位点序列,重新比对后3个可变区位点为V2(203~397)、V3(583~657)、V4(735~855)。截取出可变区后进行实际碱基数目分析,发现在V2、V3、V4可变区序列中S000583665、S000830684、S000346245、S000120585这4条序列中存在实际碱基数目较少的可变区片段。为了保证使用完全一致的物种类别构建进化树,只保留3个可变区中实际碱基数目均较高的序列,最终得到可用于构建进化树的V2、V3、V4可变区以及全长序列数据。
将用于进化树构建的数据在ClustalX中重新比对,比对完成后将ClustalX生成的.dnd文件使用TreeView软件打开,构建V2、V3、V4可变区及全长序列进化树,结果如图4~图7所示。
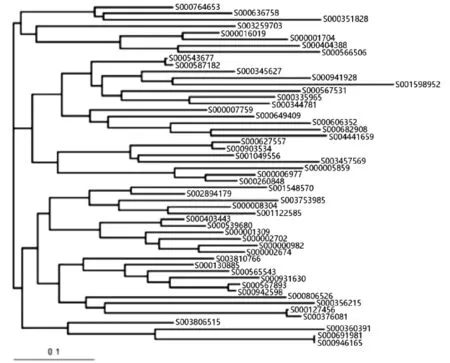
图4 V2可变区进化树

图5 V3可变区进化树
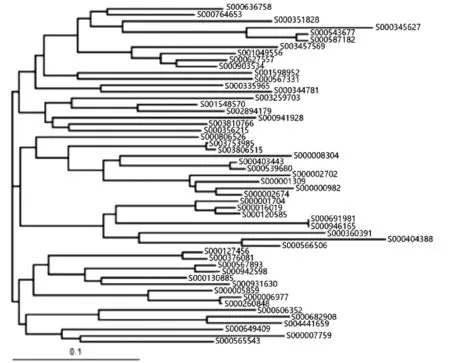
图6 V4可变区进化树

图7 全长序列进化树
结果显示,V3可变区片段生成的进化树与全长序列生成的进化树有着较大的偏差,而V2、V4可变区在进化树结构上与全长序列很相似。对于进化距离较小的序列对,使用V2、V4可变区构建进化树仍能得到比较接近全长序列的进化关系,例如S000543677与S000587182、S000691981与S000946165、S000007759与S000649409。以S000543677与S000587182为例,在4棵进化树中这两条序列之间的进化距离都非常小,将这两个序列视为一个结点,可以看出,在全长序列中与该结点进化距离最小的序列为S000345627,这三者的距离关系与在V2、V4可变区进化树中的距离关系是吻合的,而与V3可变区构建进化树的结果相距甚远。这说明3个可变区在进化关系上与全长序列之间都存在一定的差异,但V2、V4两个可变区所构建的进化树在可信度上要略优于V3可变区。
2.2 算法描述
为了定量分析可变区进化树与全长序列进化树之间的相似程度,需要建立一个能够衡量两棵进化树之间相似度的方法。因此,本文提出一种能评价相同序列不同片段构成的不同进化树之间进化关系相似度的方法,即对任意一棵进化树,都可以建立一个层次距离矩阵,通过对两棵树层次距离矩阵的比较,可以得到它们之间的距离差异值,进而可以分析进化关系的相似程度。层次距离矩阵算法示意图如图8所示。对于树中任意两个叶子结点,两两计算层次距离,结点间的层次距离定义为:若两个结点有同一父结点,则两个结点间层次距离为0;否则,层次距离为两个结点向上到达第一个共同祖先结点的距离权值。
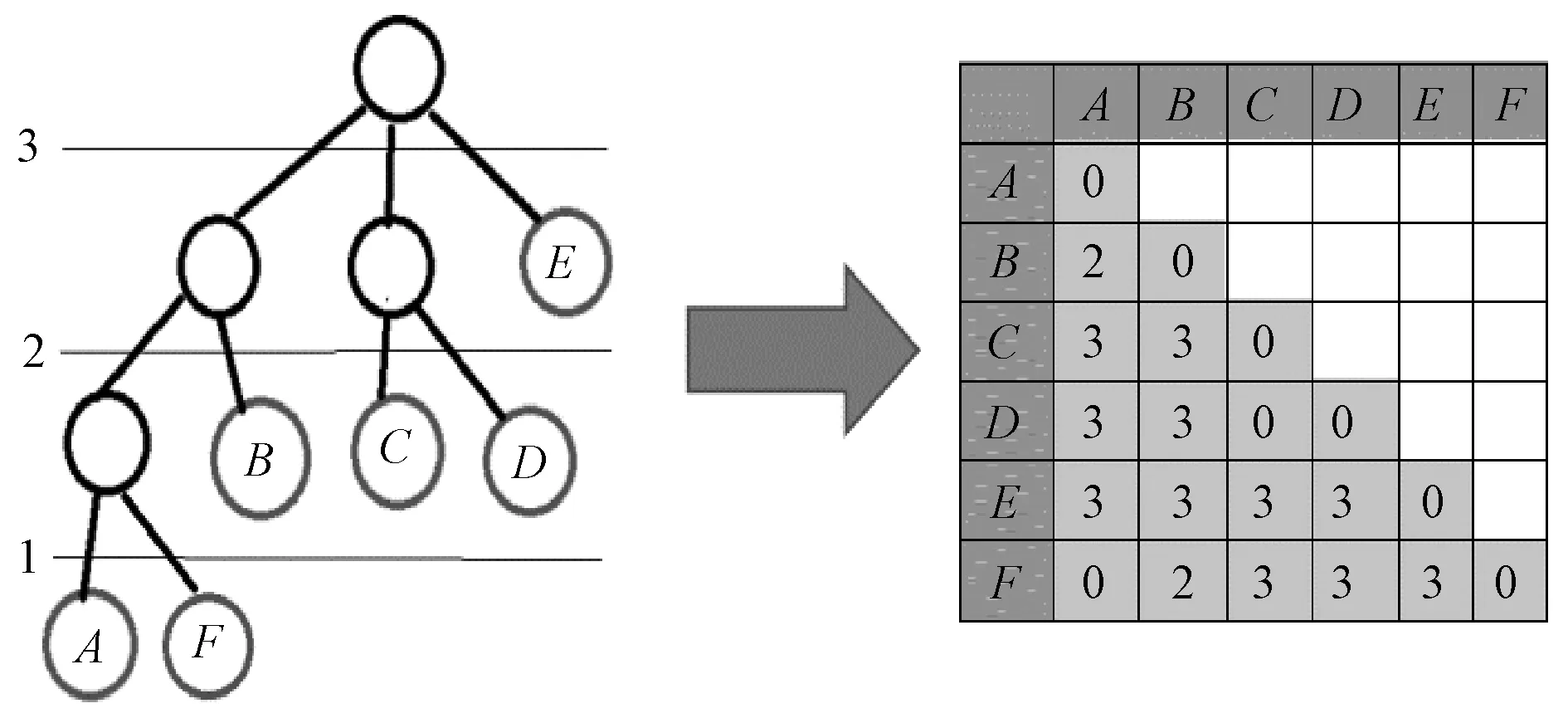
图8 层次距离矩阵算法示意图
若M、N为树中两个叶子结点,则其层次距离可以表示为
(1)
其中:LM和LN分别表示结点M和N下方路径所在层数;LMN表示结点M、N最近公共祖先结点下方路径所在层数;W表示路径上方结点的距离权值。
A、D两个叶子结点之间的最近公共祖先结点为根结点,若各层的距离权值如图8所示,则A、D两个结点间的层次距离为根结点的距离权值3。在完成了两棵树的层次距离计算后,得到任意两个序列在两棵树中层次距离的差异值,可以表示为
(2)
将两个层次距离矩阵中所有对应位置的差异值相加,则两棵树之间的进化关系相似度指标可以表示为
(3)
其中:S为所有叶子结点的集合。将可变区的层次距离矩阵与全长序列层次距离矩阵按位置相减并取绝对值,可以得到差异值矩阵,该矩阵可以较准确地反映任意两个序列在不同片段构建的进化树的差异程度。相似度指标D能一定程度地反映两棵进化树在整体上的层次距离差异。D值越大,两棵树中对应的序列对之间层次距离的差异越大,表明两棵树之间的相似度越小。
2.3 算法实现与结论
对于图4~图7中所示的进化树,将各叶子结点序列名称以数字0~52进行编号,并以大小写英文字母A~Z、a~z为中间结点编号,可得到各序列在不同树中从根结点出发的路径。在两两路径之间进行层次距离矩阵计算时,首先倒序寻找两个序列中第一个相同的中间结点,接着提取该中间结点的层次权值,得到两路径之间的层次距离。利用式(1)~(3)进行差异值矩阵计算,可得V2、V3、V4可变区与全长序列之间的距离差异值分别为59 052、87 154和45 848,相似度分别为41%、13%和55%。可以看出,V4可变区与全长序列之间两两序列的距离差异值要小于V2、V3可变区,且V4可变区与全长序列之间进化关系的相似度为55%,大于V2、V3可变区与全长序列之间进化关系的相似度。因此,V4可变区所构建的进化树更接近全长序列所构建的进化树,在可信度上要优于V2、V3可变区,在细菌物种进化关系上V4可变区较V2、V3可变区片段更为接近全长序列。
2.4 算法比较
与传统的距离与相似度算法如欧氏距离算法、马氏距离算法、汉明距离算法相比,层次距离矩阵算法的优点在于若两结点都是根结点的子结点,在不使用距离权值的情况下,两结点的层次距离是很小的,这与实际情况不符,而引入距离权值之后则可以解决这一问题,即在树中越接近顶端的结点与相同层次的结点之间的序列距离越大,两结点的层次距离差距也越大。分别使用层次距离矩阵算法、欧氏距离算法、马氏距离算法、汉明距离算法计算V2、V3、V4可变区与全长序列之间的距离差异值,结果如表2所示。这四种算法的均方值分别为21 096.02、29 887.54、28 318.72和34 960.29,精度分别为79.0%、70.2%、71.7%和65.1%。可以看出,层次距离矩阵算法与传统的距离与相似度算法相比,在各可变区与全长序列距离上的均方值更小,计算距离的精度更高,使用这一算法可以计算树中任意两个叶子结点之间的层次距离,并得到不同树之间所有序列对在层次距离上的差异值,而引入距离权值主要是基于对距离偏移大小的考虑。以上分析对物种进化来源分析研究具有重要的指导意义,若要研究某一菌种的突变来源,可利用可变区分别构建进化树并比较相似度的方法进行分析。
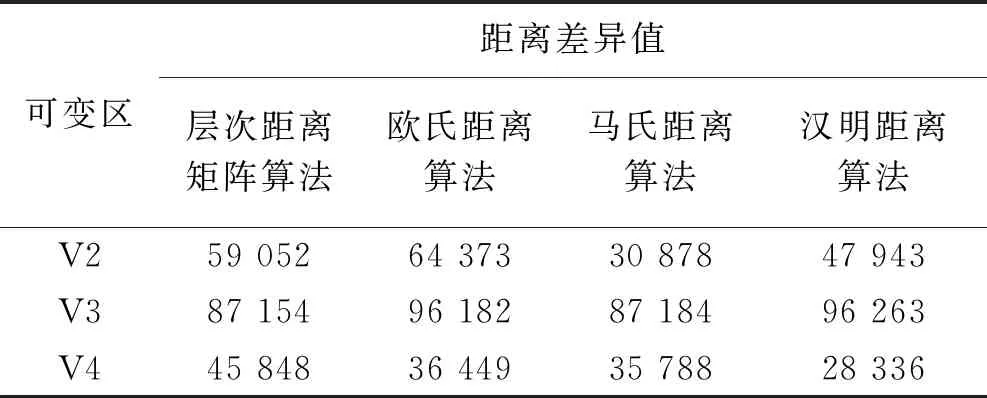
表2 不同算法计算的可变区与全长序列之间的距离差异值
3 讨论
本文以RDP所提供的细菌16S rRNA数据为基础,对不同可变区物种进化关系的相似性进行了研究,分别对这些可变区片段进行重新比对和构建进化树,并且与全长序列构建的进化树进行比较,发现V4可变区在进化关系上与全长序列更为贴近,V4可变区构建进化树的可信度要优于V2、V3可变区。本文使用的层次距离矩阵算法对物种进化来源分析研究有一定的指导意义,较传统的距离与相似度算法具有更好的性能。但是,这并不能否认单独利用可变区进行物种进化关系分析存在一定的局限性。另外,本文所提出的相似度计算方法虽然能在一定程度上反映两棵进化树之间的相似程度,但也存在一些不足之处。在提出层次距离矩阵计算方法之前,曾尝试使用树的层次遍历进行转换代价计算,在叶子结点相同的情况下,这种方法可以将一棵树转换成另一棵树的形式,通过计算这一转换过程中的代价,可以评价两棵树的相似程度,但是该方法的问题在于两棵树的非叶结点数目可能不同,如果能够解决这一问题,那么使用转移代价来评价树的相似程度是更为可行的一种方法。
