基于第一性原理对尖晶石结构LiMn2O4正极材料和相关化合物进行理论研究
2022-01-22伍澎贵姜兴涛宁玉娟黄东雪梁兴华
伍澎贵,姜兴涛,张 宇,宁玉娟,黄东雪,梁兴华
(广西科技大学 机械与汽车工程学院,广西 柳州 545006)
0 引言
由于石油、天然气等可再生能源的紧缺和环境污染不断加剧问题的出现,研究学者们逐渐把目光投向于可再生新能源材料的研究和开发,从而带动了新能源电车以及动力电池的发展[1]。锂电池作为新的能源供给方式,为新能源发展的重点[2]。目前,各研究团队对于正极材料的研究相对深入。而研究学者们现也正在寻求一种具有原材料丰富且价格便宜、对污染环境友好污染小、高能量比、高工作电压、电化学性能好、安全性高等优点的电池正极材料。因此,研究学者们找到了一种满足上述条件的尖晶石LiMn2O4材料,但由于其仍然存在诸多缺点,如:在充放电过程中比容量快速衰减、不耐高温、容易出现Jahn - Teller畸变效应,在很大程度上阻碍了它的大规模商业化发展。为此,研究学者们需要找寻一种有效且合理的改良途径去优化尖晶石LiMn2O4材料的缺点,目前主流有效的改良途径是掺杂[3]。通过在材料中掺入适当的过渡金属X原子代替某些Mn原子,可以有效地改善材料的电化学性能[4]。通过进行元素掺杂,提高了容量比、能量比使其稳定性能更好,元素的掺杂导致电位能表面的变化,可能会改变Li+迁移的屏障,还有可能改变Li+迁移的路径[5],因此对LMO材料进行深入研究Li+迁移路径显得非常有必要。
近年来,第一性原理研究在Li+电池领域占有越来越重要的地位,通过第一性原理计算可以解释材料的内部结构、材料掺杂前后属性变化等信息[6]。申海燕等利用第一性原理计算了尖晶石型锂锰氧化物的结构特征,结果表明:缺陷型LiMn2O4中理论容量随着Li含量的增加而减小;王延庆等利用第一性原理计算了Ni -3d轨道对LiMn2O4的诱导作用,结果表明:Mn位掺杂Ni后Mn - O的键强增加,提高了LMO的电子导电率以及其结构的稳定性,进一步加快了Li+的扩散速度[7]。
高农等使用温差法生成LAMO材料,并对其进行测试[8],从宏观方面解释了一定量 Al的掺杂改善了LiMn2O4电池的循环性能,但是研究学者们对于铝锰酸锂的微观结构研究较少[9]。本文根据密度泛函理论和第一性原理相结合对LMO结构模型进行一系列的分析和计算,对分别掺入Ni2+、Al3+前后LiMn2O4的总能量与晶格常数、总态密度的计算分析。通过对计算结果与实验结论的对比,表明两者的结论一致。
1 模型与方法
尖晶石LiMn2O4属于立方晶系,为Fd-3m空间群[10],晶胞结构类型如图1(a)所示。在尖晶石结构中,氧原子占据32e位置,锰原子占据16d位置,而锂位于1/8氧四面体间隙(8a)位置[11]。Li+通过空的相邻四面体和八面体间隙沿8a-16c-8a顺序路径在Mn2O4三维网络结构中脱嵌和位置穿梭[12]。
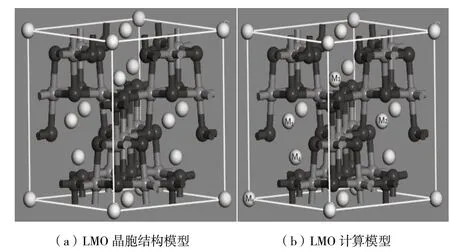
图1 LMO晶胞体图
它们沿传导通道交替排列,并在整个晶体结构中建立了三维迁移网络。一些具有较大四价阳离子的组合物,还显示出由于Li+的置换而引起的较低对称三斜晶相。在这种晶胞结构中,Li+在内部迁移按照8a-16c-8a顺序移动[13],考虑到掺杂元素对晶体结构的变化及Li+的迁移,针对掺杂位对Li+迁移的影响,我们计算M1到M2方向的迁移,在图1(b)中给出了在尖晶石中Li+的迁移路径。观察M1到M2的迁移过程中,掺杂元素对Li+的影响。
在晶体结构中离子迁移是由间隙或空位之间的离子热活化跳跃驱动的。并且,宏观扩散取决于材料的微观结构。我们已经对LiMn2O4和相关材料的菱形结构的Li+扩散进行了大量实验研究。在原子模型中研究运用密度泛函理论(DET)和动力学模拟研究LiMn2O4和LiNi0.5Mn1.5O4中的Li+迁移途径。Vi+离子的部分取代针对多种元素进行,尽管用Ge等代替Ni取代Mn元素的结构确实稳定了金属Li,但它们的生产成本明显更高,并且存在具有较大阳离子的结构中的多态性问题。因此,与未取代的LMO相比,DFT研究集中于阳离子取代对晶体结构和能垒的影响。在以下各节中,描述了尖晶石结构模型和计算DFT设置,给出并讨论了结果,最后给出了结论。
(1)LMO的结构模型。为了进行计算,使用了如图1(a)所示的具有56个原子的尖晶石结构的常规六面体晶胞。除去一个Li原子以产生扩散所需的空位。作为参考结构,选择了未取代的LMO组合物,对其余两种阳离子(X=Ni,Al)取代Mn的LXMO组成进行了计算。对于每种组合物进行了晶胞体积和原子位置的弛豫。通过用晶胞LXMO中的X阳离子仅置换16个Mn原子之一,已经描述了较低浓度的取代元素对扩散路径的直接影响。对于这些结构,仅原子位置已被弛豫,LMO的弛豫单元常数也已用于所有的LXMO,因为通过仅替换晶胞中的16个Mn原子之一,体积变化很小。
(2)几何结构优化。本文采用Materials Studio软件,在CASTEP中的Geometry Optimization模块进行建模与能量计算,该模块基于密度泛函理论对LMO与LXMO晶胞的几何结构进行研究,计算中采用广义梯度近似(GGA-PBE) 方案来描述电子与电子间的交换关联能[14]。
(3)锂空位和空位扩散。通过对Li+分布和晶体结构的对称性的分析,我们得出了晶格中两个独立的锂扩散路径,它们将不同平面中的Li位点相互连接,并且因此,尖晶石结构中形成三维扩散网络。然后通过NEB方法优化Li+迁移路径,并且计算出迁移能垒。通过中子衍射(ND)实验推导了LMO中Li优先占据M1位。LMO在温室下显示出Li阳离子原理M1位的无序现象,无法用K=0时的DFT计算来描述。首先假定空位介质的Li扩散。
(4)计算方法。计算使用CASTEP中的Energy计算模块进行理论计算,计算参数设定为:原子间相互作用力的收敛标准设置为0.02eV/Ang,平面波截止为500eV,设置网格参数为4×4×4。原子最大位移收敛标准设置为1.0×10-5µeV。优化后数值与原始实验设定晶胞常数a相比存在误差,实际误差为1.8%。通过使总能量最小化来完成晶胞体积的松弛,并且使原子位置松弛直至作用在原子上的剩余力小于10-3eV/。通过微动弹性带(NEB)方法获得相邻M1个位点之间锂跃迁的最小能量路径(MEP)。作用于插值反应路径的NEB图像上的总力阈值设置为0.05eV/。
2 分析与讨论
2.1 结构优化与总能量
根据总能量最低原理对三种模型优化处理,得到如表1所示优化后模型晶格参数和总能量。为了便于计算,节省计算时间,选用5×5×5模型进行计算。从图 2(a)、(b)、(c)三种晶胞结构元素键可以看出它们都是Fd-3m对称性的立方晶体。与LMO晶格常数初始值(a=8.221(4))对比,LMO结构优化后,原胞体积V=0.5609nm3,晶格常数减小,误差仅为1.56%,在赝势平面波方法所允许的范围内,说明模型建立正确、可靠。与未掺杂相比,在Mn位掺杂Ni2+后导致晶格常数增大,原胞体积增大为0.5641nm3,总能量的变化量为-1152eV。而Mn位掺杂Al3+后导致晶格常数减小,原胞体积减小为0.5578nm3,总能量的变化量为-538eV。

图2 (a)LMO、(b)LAMO 和(c)LNMO 三种晶胞结构元素键
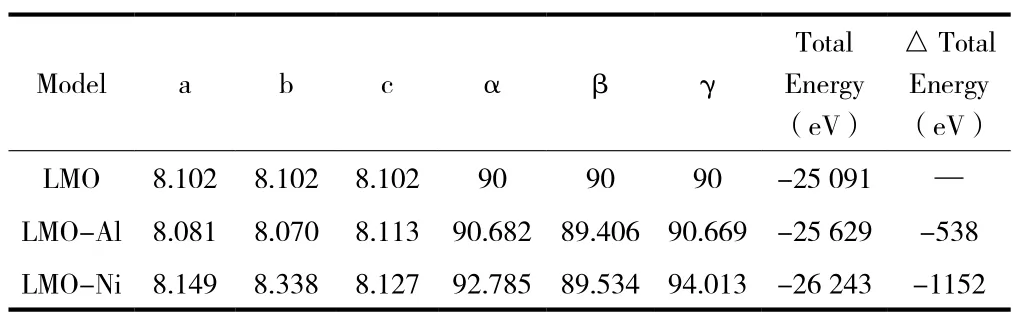
表1 优化后模型晶格常数和总能量
综上所述,经过Mn位掺杂Ni2+、Al3+后体系能量的降低,说明晶体结构的稳定性提高。在Li+迁移时,结构不易会发生改变,能够提高材料的循环稳定性和使用寿命。因此,在Mn位掺杂Ni2+、Al3+,均可以提高LMO结构的稳定性,且Mn位掺杂Ni2+的稳定性更好,与文献[16]报道吻合。而Mn位掺杂元素的不同导致晶格常数变化不同,这可能与晶体结构、掺杂元素的浓度及掺杂位置有关。
2.2 总态密度与分态密度的分析
在第一性原理计算中,对几种材料进行了晶胞结构优化。然后,分别得到图3LMO、LAMO和LNMO的总态密度和分态密度图,横坐标为能量,纵坐标为态密度。通过元素掺杂后,元素之间的分布较为接近。这表明掺杂元素后,LMO尖晶石结构未发生结构改变,保留的尖晶石结构的稳定性。从图3可以看出,在元素掺杂后,Mn与O的电子轨道发生轻微变化,表明材料的金属性能未发生改变。
通过分析图3可知,未掺杂与分别掺杂Al3+、Ni2+的三种材料在-7~1eV,随着元素的替换,LAMO费米能级附近的峰主要由Al-p、O-s与Mn-d电子轨道进行贡献,Mn、O元素的电子轨迹明显重叠,由此说明Mn和Ni原子的加入,使得Mn与O之间的电子轨道明显重叠,并且元素的替换,提高了元素间的相互作用,稳定了晶胞结构。在-20eV附近的电子轨道密度峰十分尖锐,说明成键作用较小,反键作用强烈,局域化明显。由于Al的半径小于Mn,原始Mn-O的键长缩短,这有利于提升Mn-O骨架的结构稳定性。在-40eV附近观察看来,主要由Li-1s轨道提供,该峰的面积积分为2,表明该峰完全由Li-1s电子轨道中的两个电子提供,该峰比较尖锐,且总态密度在费米能附近的峰强无Li的贡献,说明Li与Mn-O作用很弱,Li可以在Mn-O结构中自由独立地穿梭脱嵌,元素的掺杂导致材料性能提升。在Ni元素掺杂LMO晶胞结构后,使结构出现了O-2p能带,导致在掺杂位出现Mn与O元素在电子轨道上的成键能力增强,这与文献[17]的实验结果一致。

图3 (a)LMO、(b)LAMO和(c)LNMO三种晶胞结构能量分布图

2.3 晶胞结构的体相迁移
在晶胞结构图中有三条迁移路径由图1(b)所示,分别为 M1-M2、M1-M3、M4-M5,对于迁移路径的选择,仅考虑掺杂元素对Li+迁移的影响,一方面是Li+的迁移通道,另一方面是局部的势能面,这两个因素相竞争,所以最终决定在掺杂位附近选取M1-M2两个位置的Li+迁移情况进行理论计算。分别计算出M1-M2两个位点的对向迁移能垒,得出迁移能垒相同,为0.74eV。之后首先计算出未掺杂位置锰原子的迁移能垒对Li+迁移的影响,在图4(a)中可以看到在LMO晶胞结构中M1-M2迁移路径,然后建立两个晶胞结构,命名为生成物与反应物,接着计算间隙迁移需要的迁移能垒为0.72eV。在图4(b)、(c)中我们可以看到经过Al与Ni原子替换后LMO晶胞结构中M1-M2的迁移路径,对M1-M2两个位置迁移都需要的能量,经过计算得出结果分别为0.32eV和0.57eV。由此得出结论:替换后的原子使得附近Li+迁移更加容易,Li+在Mn原子束缚下,距离近时受到Mn原子的影响比较大,而Mn原子经过Ni与Al原子替换后,键长变长,使得晶格变大,对Li影响较小,使其容易在晶胞结构中穿梭。
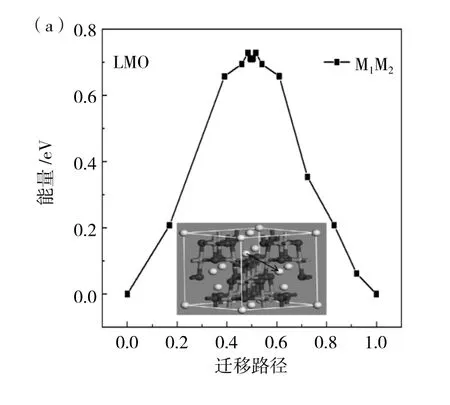
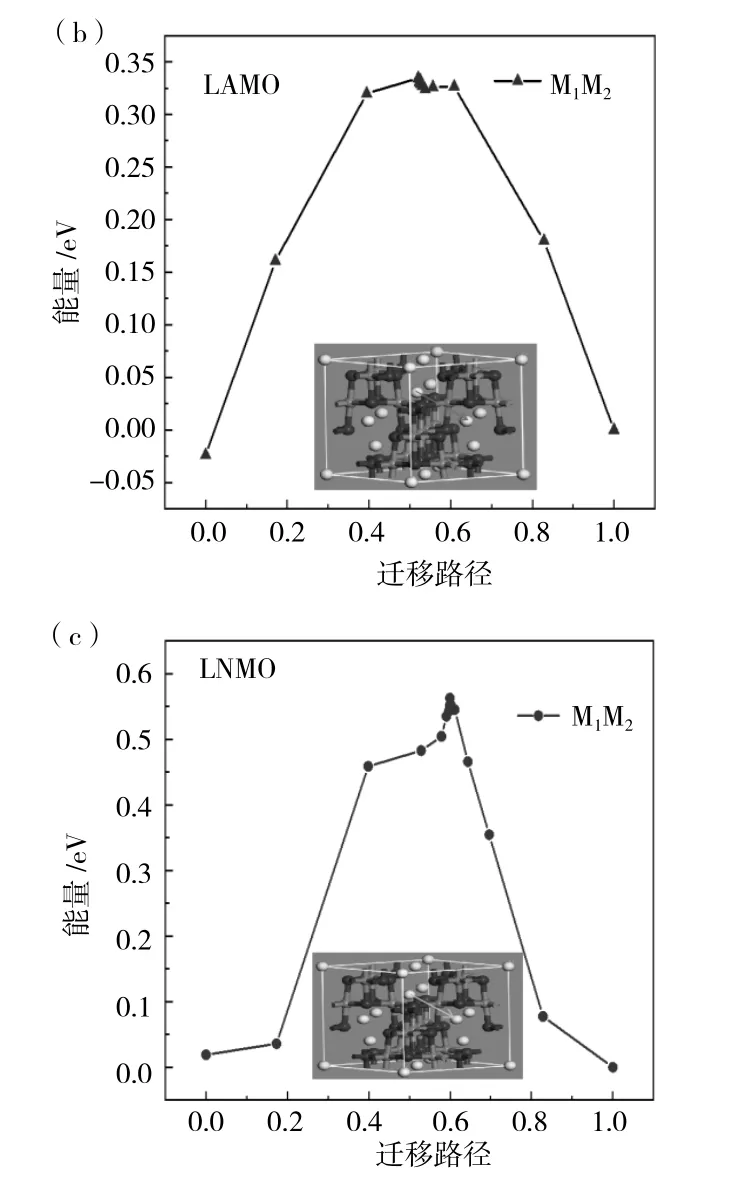
图4 (a)LMO、(b)LAMO和(c)LNMO三种材料的迁移路径能垒图
3 结论
本文根据密度泛函理论以及第一性原理相结合对LMO结构模型进行一系列的分析和计算。通过结构分析可以看出,Mn位掺杂Ni2+、Al3+后LMO体系结构更稳定,且Mn位掺杂Ni2+时总能量的变化量为-1152 eV,稳定性更高。由态密度图可以看出,在Mn位掺杂Al3+后,由于原始Mn-O的键长缩短,这有利于提升Mn-O骨架的结构稳定性,同时加快了LAMO晶胞结构中Li+的迁移速率。在LMO晶胞结构中,经过Ni元素替换Mn元素,由于嵌入了Ni-3d电子轨道,诱导LNMO增加了O元素的2p电子轨道,由于晶胞结构中Li+迁移所需要的能量大部分由O-2p能带结构提供,因此,增加了尖晶石结构的导电性能。通过元素掺杂,使掺杂位键长变大。通过元素掺杂后,计算掺杂位置的锂位迁移,计算出迁移能垒,由计算可知,经过元素替代Mn2+离子,掺杂后的过渡态能量能量降低,迁移势垒更小。
