皮下脂膜炎样T 细胞淋巴瘤1 例
2022-01-20梅爱华刘劲松董亚琦
梅爱华 刘 菡 刘劲松 朱 红 武 伦 董亚琦
1.湖北医药学院附属国药东风总医院皮肤科,湖北十堰 442008;2.湖北医药学院附属国药东风总医院实验中心,湖北十堰 442008
皮下脂膜炎样T 细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma,SPTCL)是一种罕见的原发性皮肤恶性肿瘤,归属于成熟T 细胞和NK 细胞肿瘤中的一种,主要浸润皮下脂肪组织,很少累及皮肤以外的淋巴组织和器官[1],世界卫生组织于2005 年,将其正式定义为皮下原发的细胞毒性T 细胞淋巴瘤[2],目前发病机制和致病因素尚不清楚,有少数学者认为SPTCL 的发病和患者感染EB 病毒有关[3],但在后继研究中发现两者之间又不具有明显的相关性[4-5]。根据最新研究报道,有学者认为SPTCL 的发病与患者免疫系统疾病有关,推测是SPTCL 的易感条件;基因HAVCR2 突变,促使部分T 细胞免疫球蛋白黏蛋白-3(T-cell immunoglobulin mucin-3,TIM-3)功能损害,而TIM-3 是T 细胞亚群表面特异性检测的生物标志物,可以抑制T 细胞功能,从而使SPTCL 患者体内促炎症细胞因子表达水平升高[6-7],这些基因的突变,可通过免疫调节以及表观遗传和信号转导调控参与SPTCL 的发生发展。SPTCL 临床症状不典型,可通过影像学CT 和MRI 检查、病理学、免疫指标检测进行诊断和鉴别诊断[8]。目前,缺乏特异性的治疗方案,若早期确诊和治疗,可获得相对满意的治疗效果。湖北医药学院附属国药东风总医院(以下简称“我院”)于2019 年1 月收治了1 例SPTCL 患者,现报道如下:
1 病例资料
患者,男,72 岁,因“右小腿起疹20 余天”于2019 年1 月收入我院治疗。患者20 余天前无明显诱因右小腿出现紫红斑,约4 cm×4 cm 大小,压之有疼痛,当时未予重视,未予特殊诊治,起病过程中伴有间断发热,体温最高37.9℃左右,一般晚上8:00-9:00 体温最高,发热前自感发冷,无明显畏寒、寒战,伴有咳嗽、咳白黏痰,有明显乏力、胸闷、活动后气促,伴有双下肢水肿,右侧明显,右侧胸壁、面部稍肿胀,无端坐呼吸,无夜间阵发呼吸困难,无肌肉疼痛、关节疼痛,无明显腹痛、腹胀、腹泻,无尿急、尿频、尿痛,发病以来曾查下肢动静脉彩超未见异常,肝肾功基本正常(谷丙转氨酶为31 U/L、谷草转氨酶为62 U/L),血常规提示有轻度贫血(81.27 g/L),今为求进一步诊治,遂入我院皮肤科,皮肤科门诊以“皮疹查因”收治入院。
起病以来,患者精神、睡眠、食纳一般,大小便正常,体力体重无明显变化。既往史:有颈椎病史多年,1996 年患肩周炎,1999 年行前列腺电 切术,2000 年摔伤致左侧后肋骨折,1 年前曾发现右侧胸壁肿胀,胸部CT 考虑右侧胸壁炎,给予局部外用药抗炎治疗。
体格检查:T 37.1℃,P 75 次/min,R 20 次/min,BP 122/77 mmHg(1 mmHg=0.133 kPa),神清,步入病房,查体合作,全身皮肤黏膜无黄染,左侧腹股沟可触及一花生米大小的肿大淋巴结,质地中等,移动度欠佳,双肺呼吸音稍粗,双肺呼气相均可闻及哮鸣音,右下肺呼吸音稍低,心率75 次/min,律齐,各瓣膜听诊区未闻及杂音,腹软,无压痛反跳痛,肝脾肋下未及,双肾区无叩痛,双下肢水肿,右侧明显,右侧胸壁、面部稍肿胀,病理征未引出。皮肤科情况:右小腿见一约4 cm×4 cm 大小紫红色斑片、斑块,轻压痛,其上可见点片状糜烂面及少量淡黄色渗夜(图1)。病理诊断结果见图2A~B。免疫组织化学染色结果示:κ(-),λ(-),CD3(+),CD5(个别细胞+),CD43(++),CD20(-),CD79α(-),Pax5(-),CD21(-),CD23(-),CD45(++),CD56(+),CD68(+),CD4(+),CD8(-),CD30(-),ALK(-),EMA(-),Ki-67(局灶30%),CKpan(-),VIM(+),HMB45(-),TiA-1(+),TdT(+)(图2C~F)。原位杂交结果:EBER(+/-)。根据免疫组织化学染色及原位杂交结果,病理诊断如下:镜下见均为T 细胞淋巴瘤(图2A~B),考虑为皮下脂膜炎样T 细胞淋巴瘤。
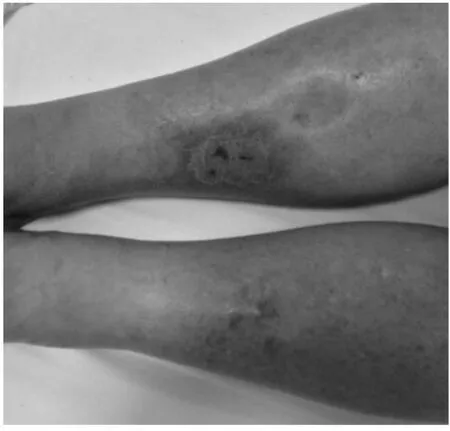
图1 右小腿可见明显病变部位

图2 部分病理检查及免疫组织化学染色结果(100×)
结合上述症状、体征和实验室检查,诊断为皮下脂膜炎样T 细胞淋巴瘤。建议行化疗,患者及家属拒绝,治疗上根据药敏给予抗感染(万古霉素、哌拉西林/他唑巴坦)、输白蛋白、护胃补钙、利尿补钾及小剂量甲强龙、甲氨蝶呤等控制病情。2 个月后死亡。
2 讨论
SPTCL 是一种细胞毒T 细胞的淋巴瘤,占所有非霍奇金淋巴瘤的1%以下,属于少见类型的淋巴瘤[9],在白种人中少见[10-11],但在亚洲人中常见[12-13]。发病无明显性别差异,各年龄段均可发病,平均发病年龄为43 岁[14]。临床首发表现常为非特异性皮下结节,不警惕容易误诊,皮损常见于四肢和躯干,还可见于面、颈、腋窝、腹股沟和臀部[15],主要表现为黄褐至红色的皮下结节或斑块,大小差异很多,其直径0.5~9.0 cm不等,无压痛,早期无明显淋巴结受累,溃疡和皮外病变少见,其中约1/3 的患者可能出现噬血细胞综合征,表现为持续性高热,肝、脾肿大,全血细胞减少,凝血功能异常[16]。
病理检查是确诊SCPTL 最主要的方法[17],其特征是皮下脂肪组织内有原发的小、中或大的多形性T 细胞,围绕脂肪细胞呈花环状排列,瘤细胞有明显的核碎裂、核坏死[18]。肿瘤组织中有组织细胞吞噬红细胞和坏死碎屑后形成的豆袋状细胞,以及反应性组织细胞,常可合并有脂肪坏死、凝固性坏死,特别是损害较大时,可见广泛脂肪坏死,坏死脂肪常导致组织细胞反应,包括多核巨细胞或肉芽肿样增生[19]。SPTCL 瘤细胞的侵袭性较强,病情进展非常迅速,自然病程凶险,但患者对全身联合化疗如环磷酰胺联合多柔比星、长春新碱和泼尼松方案(CHOP 方案)及局部放疗较敏感,治疗有效[20],经CHOP 或CHOP 样方案化疗后,平均随访时间24 个月,中位生存期为27 个月[21]。本例患者由于家属拒绝治疗,预后不好。
SPTCL 是一类特殊类型的T 细胞淋巴瘤,确诊的金标准仍然依靠病理,发病机制仍不明确,初始症状缺乏特异性[22],因此CT、MRI 鉴别诊断尤为重要[23],尽量减少误诊与漏诊的发生率。治疗方面目前尚未统一,联合用药的治疗模式是目前治疗复发难治性SPTCL 患者的趋势[24]。对TIM-3 功能缺陷的SPTCL 患者,免疫抑制剂起到了较好的疗效[18]。也有研究表明同种异体造血干细胞移植治疗难治性SPTCL 是合理的[25-26]。这些研究,仍需要大量样本、多中心研究,需要进一步的临床观察。制订规范、标准化的治疗方案是临床上亟待解决的问题。
