B4C(0001)及Al-B4C(0001)表面的结构和电子性质
2022-01-14赵玉文张航舶
赵玉文,张航舶,朱 岩
(河北科技师范学院物理系,河北 秦皇岛,066004)
碳化硼(B4C)是一种重要的非氧化物陶瓷,与其他的陶瓷相比,其最为突出的就是它同时具有超高硬度和低密度这2种特点[1,2]。室温时,B4C的硬度仅次于金刚石和立方氮化硼的硬度,而高温时,它的硬度超过了金刚石和立方氮化硼[3]。此外,B4C的密度只有2.52 g/cm3,仅为钢铁的1/3。这2个特点是其他材料无法比拟的。因此被广泛用于海洋船舶、装甲防弹、耐火材料、航空航天、核反应堆的屏蔽材料等领域。但由于B4C强的共价键使其晶界滑移阻力很大,导致其很难烧结致密,这极大地阻碍它的应用。因此,大量的研究集中在通过提高烧结工艺,或采用多相复合的方法解决碳化硼烧结温度过高、韧性差的缺点[4~6]。目前,性能比较优异且实际应用的为Al/B4C基复合材料,金属Al具有好的韧性,耐低温等优点,而且密度与B4C相近,因此该材料兼具Al和B4C的优点。为了确定Al/B4C复合材料的微观结构和性质之间的关系,一些研究人员通过X射线衍射和扫描电镜等方法对Al/B4C界面的微观结构进行了研究[7]。但由于其结构复杂,目前实验技术还无法对其界面的精密结构和电子性质进行精准的测量。而材料的精确结构直接与其性质及应用息息相关,明确其结构十分必要。一些课题组试图从理论上揭露其真实结构,但大多研究集中在体B4C材料[8~10],虽然Xian等[11]研究了Al与B4C界面的结构,但其构建的B4C表面模型为非化学计量比,与实际中存在一定差距。因此,本次研究通过第一性原理方法,从理论上系统地研究B4C表面及Al原子吸附在B4C表面的结构及其电子性质。
1 计算方法
本次研究采用基于密度泛函理论的程序包(Ab-initio Vienna Package Simulation,VASP)来进行的[12]。采用了投影缀加平面波(Projector Augmented Wave,PAW)方法[13],描述原子实与价电子之间的相互作用。分别用3s23p1,2s22p1和2s22p2的价电子构型生成了Al,B和C原子的赝势。利用Perdex Burke Emzerhof (PBE)形式中的广义梯度近似(GGA)处理交换相关能量函数[14]。收敛标准设定为:体系总能量变化小于1.0×10-6和每个原子受力小于0.001 eV/nm。选取的平面波截断能Ecut为600 eV,k点为0.003/nm。对于表面结构,构建了厚度为1.5 nm的真空层,以确保上下两层表面之间的作用力可以忽略。
2 结果与讨论
2.1 体相B4C
B4C为菱形对称结构,空间点群为R3m,1个菱形的单元格里有15个原子,其中有1个由12个原子构成的二十面体,和位于菱面体中心的1个沿[111]方向排列的3个原子构成的线性链(图1(a))。虽然目前的实验技术无法直接测得B,C的精确占位,但根据已报道的理论工作与核磁振动谱实验相结合[8],推导出1个C原子位于二十面体的极性位置,二十面体的其他点阵由B原子构成,沿着[111]方向以C-B-C构成线性原子链。因此笔者采用基于(B11Cp)+(C-B-C)单元胞构型建立的六方结构,原子最密集排列为(0001)面(图1(b))。为了验证所建模型的准确性,计算了B4C的生成焓,其公式为:

图1 B4C的晶体结构(a)菱形晶胞,包括1个二十面体和1个线性链;(b)本工作中使用的六方体结构C原子(黑色球)位于线性链的两端和二十面体的极性位置;B原子(灰色球)位于线性链的中间和二十面体的位置
(1)

2.2 B4C(0001)表面
B4C的原子最紧密堆积面为(0001)面,也是最易暴露面,因此笔者对B4C(0001)结构进行系统地研究,采用45个原子的模型(其中B原子36个,C原子9个,符合化学计量比)。笔者以表面终止元素分类,定义了4种表面,分别为以1个C原子(C-终止);1个B原子(B-终止);3个B原子(BBB-终止);2个B原子加1个C原子终止(BBC-终止)。将4种结构优化并计算其表面自由能,寻找出最稳定的表面结构。表面自由能公式为:
(2)
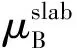
(3)
将公式(3)代入到公式(2)中:
(4)
此外,表面中每个种类的化学势应该小于其体相阶段的化学势:
(5)
否则,化合物分解成元素相是不稳定的。考虑到生成焓的定义式公式(1),可以推导出以下不等式关系:
(6)
公式(6)与公式(4)结合,预测表面自由能的范围[16,17]。


图2 C-终止,B-终止,BBB-终止和BBC-终止C(0001)表面的表面自由能与的关系
不同原子的终止面的态密度在费米能级附近的差异较大。由于表面的表面态的存在,4个结构在费米能级附近均有电子分布。C-终止的费米能级所对应的DOS值最小,其结构相对稳定(图3)。通过计算C-终端B4C的投影态密度,可知主要是B的p轨道电子与C的p轨道电子在费米能级附近有比较强的相互作用,其峰值在费米能级以下(图4)。

图3 4种表面结构的态密度 图4 C-终端B4C的投影态密度 (垂直虚线表示费米能级) (垂直虚线表示费米能级)
通过表面自由能与态密度图,结合分析得出C-终止的B4C(0001)表面是最稳定的。因此下面的工作选择C-终止的B4C(0001)表面为研究对象,研究Al吸附在B4C(0001)表面的结构和电子性能。
2.3 Al-B4C(0001)结构
本次研究将Al吸附在C-终止B4C(0001)表面的上,为了确定Al原子吸附在C-终止B4C(0001)表面的最佳位置,笔者吸附了3种不同的位置(图5),分别称为顶位,空位和桥位。顶位是将Al原子直接放置在C原子的上方;空位表明将Al原子放置在C-终止B4C(0001)表面第一层的空心位置;桥位表明将Al原子放置在C-终止B4C(0001)表面的桥位上。
为了寻找可能的吸附位置,笔者计算吸附能Wad,其定义为[14]:
(9)
A为表面积,EAl/B4C是Al/B4C的总能量。EAl和EB4C是同一空间格子中孤立的Al和B4C表面的总能量。
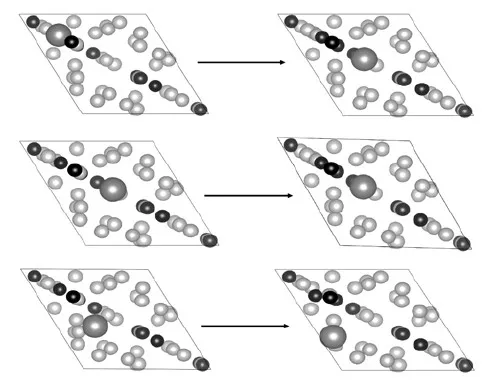
Al,B,C原子分别显示为大灰色球、灰色球和黑色球图5 Al吸附在C-终止B4C(0001)表面的3种吸附位置
通过不同吸附位置的吸附能的结构顶位(Top-site)和空位(Hollow-site)的吸附能相同(表1)。原因是吸附在顶位的Al原子经过充分弛豫后,寻找到的能量最低的位置同样是空位,表明空位吸附的结构是最稳定的,Al最易吸附在空位(图5)。但桥位的吸附能与空位的差距只有0.24 eV,其差距很小。说明在某些特定的条件下(如高温,或存在应变),Al原子也可能吸附到桥位。

表1 3种吸附位置的吸附能
为了进一步了解Al与B,C原子的作用,分别计算空位和桥位2种吸附位置的电子结构,Al吸附在C-终端B4C表面的空位的总电子态密度在费米能级较低。空位的总的态密度主要是由C的p轨道及Al的s,p轨道共同作用的;而桥位的主要是由B的p轨道及Al的s,p轨道共同作用的。其中值得注意的是,Al原子在费米能级以下1 eV时的电子态密度,2种位置均出现峰值,但其贡献的电子轨道恰好相反,而费米能级处Al的s,p轨道贡献是相当的。吸附Al原子的结构与未吸附的结构相对比,空位的C原子的p轨道电子在费米能级附近的电子态密度变化较大,而桥位的相对较小(图6)。结果表明,空位吸附时,Al原子与C原子更易成键。从电子性质分析空位吸附为最稳定的吸附结构。

图6 不同吸附位置的投影态密度 (a)空位,(b)桥位;垂直的虚线表示费米能级的位置
为了更直观的表示Al与B4C表面原子的相互作用,利用bader电荷分析方法,计算出Al与B4C表面原子的电荷转移量。结果表明,在2种结构中Al原子都是失去电子(表2);表面的C原子获得电子,其中C原子获得的电子一部分是B原子贡献的;而处于极性位置B原子其电荷转移较少,非极性位置的B原子电荷失去电子较多。总体来说,2种结构的电荷转移差别较小,而空位吸附结构中Al与表面B,C原子的相互作用略强,这与态密度分析是一致的。

表2 Al与B4C表面原子的电荷转移量(e)
3 结 论
通过第一性原理计算,研究了B4C(0001)表面及Al原子吸附B4C(0001)表面的结构及电子性质。首先,计算了体相B4C的生成焓,结果与实验值较吻合。其次,构造了4种不同终止的B4C(0001)表面,通过对表面自由能及电子性质的分析,预测C-终止B4C(0001)表面是最稳定的。进一步研究了C-终止的B4C(0001)表面为上吸附Al原子的结构和电子性能。通过计算吸附能和电子结构,比较了3种吸附位置。笔者的计算结果表明,Al原子在B4C(0001)的空位吸附为最佳吸附位置。
