碱性膜电解水制氢技术现状与展望
2022-01-10王培灿万磊徐子昂许琴王保国
王培灿,万磊,徐子昂,许琴,王保国
(清华大学化学工程系,北京 100084)
引 言
能源作为人类社会进步的重要推动力,具有举足轻重的地位。长期以来人们一直使用传统化石能源作为能源主体,随之也带来了一些严峻问题。一方面传统化石能源作为不可再生资源,无法满足人类社会的可持续发展;另一方面,由于过度使用化石燃料排放温室气体对全球生态环境造成恶劣影响。所以,发展清洁高效的可再生能源技术,实现人类可持续发展,已经成为世界各国的共识。能源转型是实现经济社会可持续发展的必然选择,提升可再生能源比例,大力推动风电、光伏发展,是实现能源绿色转型和低碳发展的重要举措。但风电、光伏存在不连续和不稳定的问题,不能够直接并入电网使用。为了将分散的低密度能源利用起来,发展大规模电能转化与存储技术具有战略价值。
氢气作为新一代可持续利用的清洁能源载体,其优势在于:(1)氢和电能之间通过电解水技术可实现高效相互转换;(2)质量能量密度高;(3)有望在电网规模进行能源管理应用。国务院于2020年12月发布的《新时代的中国能源发展》白皮书,将发展氢能产业链纳入国家战略。将“弃风”“弃光”转化为氢能储存和利用,成为切实可行的技术路线,有利于推动国家能源转型。因此,发展温室气体“零排放”的电解水制氢技术,支撑可再生能源良性发展,成为电网级别大规模能源转化的必然选择。
1 电解水制氢技术
碱性溶液电解水(alkaline water electrolysis,AWE)和质子/阴离子交换膜电解水(proton/anion exchange membrane water electrolysis)是目前电解水的主要方式(表1)。目前发展最成熟的电解水制氢工艺是碱性溶液电解水技术,其具有操作简便的优势,已经实现了大规模商业化运用[1]。然而,AWE长期存在着由于电极与隔膜间隔较远,导致整个电解槽体积巨大,因此电解性能低(2.0 V电压下电流密度仅有约300mA·cm-2),无法满足工业需求的问题。另外,碱性水溶液电解响应速度慢,使其与间断性可再生能源耦合困难;高浓度的碱性溶液为电解液对装备要求严苛,显著地增大了制氢设备成本[2]。为此,在后续发展中,一种新型电解槽结构——质子交换膜电解水装置(PEMWE)被提出并被深入研究。
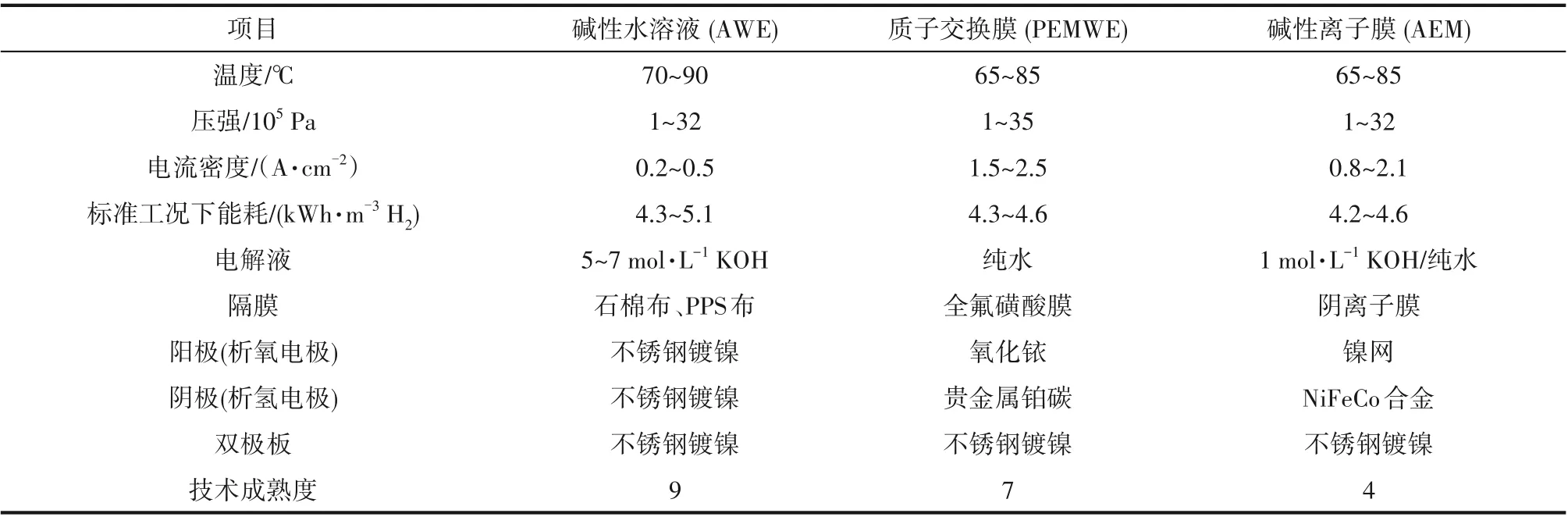
表1 电解水制氢主要技术特性比较Table1 Main characters of hydrogen production technologies by water electrolysis
图1为PEMWE的结构示意图,其中最为关键的结构为膜电极[3]。膜电极包括气体扩散层(GDL)、催化层和质子交换膜三部分,气体扩散层和催化层分别在膜两侧形成“三明治”结构。目前,PEMWE使用全氟磺酸膜(perfluorinated sulfonic acid membrane)作为阳离子交换膜(proton exchange membrane,PEM),该PEM具有极高的质子传导率。PEMWE通入纯水,以PEM为电解质,可以在高电流密度下运作,并且能够迅速作出动态响应。质子交换膜电解水与传统碱性溶液电解水相比具有启停速度快,能量利用效率高、气体纯度高、绿色环保、能耗低、无碱液、体积小、安全可靠、可实现更高的产气压力等优势。但是,PEMWE电解过程中产生的强酸性环境对电极及电极催化剂提出了较高的要求。碱性环境中使用的普通催化剂(Fe-Ni等)在酸性条件下不能稳定存在,PEMWE只能使用贵金属催化剂(Pt作为阴极催化剂,IrO2作为阳极催化剂),PEMWE的成本大幅上升,阻碍了质子交换膜电解水的大规模应用[4]。
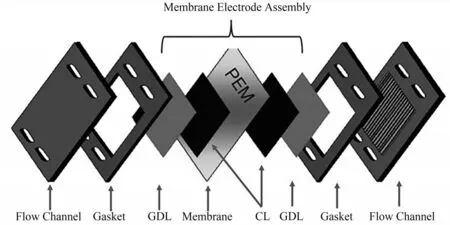
图1 PEMWE电解槽结构和膜电极组成示意图[3]Fig.1 Schematic of PEMWE electrolyzer and membrane electrode assembly[3]
阴离子交换膜电解水(anion exchange membrane water electrolysis,AEMWE)结合了AWE低成本的优势和PEMWE高效率和高便捷性的优势,是最新发展的电解水技术。阴离子交换膜电解可在碱性条件下使用非贵金属为电极催化剂,可显著地降低制氢成本。另外,阴离子交换膜电解水可使用纯水或低浓度碱性水溶液为电解液,缓解了强碱性溶液对设备的腐蚀。同时,阴离子交换膜电解水不需要使用昂贵的全氟磺酸膜,可以进一步降低材料成本。但是,目前AEMWE仍处在发展阶段,仍然存在很多亟待解决的问题[5]。例如,阴离子交换膜(anion exchange membrane,AEM)在强碱性条件下离子电导率与耐碱稳定性、力学性能难以兼得。AEM作为AEMWE中重要组成部分,它的作用是将OH-从阴极传导到阳极,同时阻隔气体和电子在电极间直接传递。但AEMWE在工作过程中,膜表面形成的局部强碱性环境使得AEM在OH-的攻击下发生降解,由此引发的膜穿孔会造成电池短路,使得AEMWE不能够长时间运行,因此发展高离子电导率与强耐碱的阴离子交换膜是进一步发展阴离子交换膜电解水的关键技术难题。
综合以上讨论,AEMWE因其独特的低成本、高效率优势被认为是最具发展前景的电解水制氢技术,但也存在一些关键技术难题比如强耐碱性膜材料等需要进一步探索解决。为了满足AEMWE技术的发展,亟需发展高效催化剂、聚合物膜、膜电极等关键材料。本文将综述电解水用催化电极、膜材料、膜电极的研究进展,并进一步阐明电解水制氢技术的发展方向,即推动AEMWE技术发展,提出其在各个关键核心部件未来研究的技术途径。
2 自支撑催化电极
电解水过程涉及两个半反应:析氢反应和析氧反应[6-8]。由于析氢过程和析氧过程是反应动力学惰性的,为此将导致高析氢和析氧过电位,显著增加电解水能耗。目前,铂系贵金属被认为是最佳的析氢催化电极,IrO2等贵金属氧化物被认为具有高析氧活性,但是这类贵金属催化剂地球含量低、价格昂贵的特点大大限制了它们的商业化的使用。为此,研究开发高性能、低成本、高稳定的催化剂对推动电解水技术的发展至关重要。
一般来说,析氢和析氧两个过程发生在电解液-气体-催化剂的三相界面处。如在析氧过程中,电子从基底表面传递到催化活性位点,同时氢氧根从溶液体相中吸附到催化活性位点处,接着发生一系列表面反应,包括电荷转移、分子重构、断键、成键等,而后吸附的氧分子从催化表面脱附。为此,通过优化调节传质过程、电荷转移过程、表面反应将能够设计得到高性能的催化剂。
近十年来,一种新型的催化电极受到了广泛的研究——自支撑催化电极。顾名思义,即将催化活性物质直接原位生长在导电基底上。如图2所示,相比于传统的粉末状催化电极,自支撑催化电极具有以下优势:①能够有效避免黏结剂的使用,从而有效地加强传质过程,显著减小电解液-电极间的电阻,同时能够暴露更多的活性位点[9-10];②实现催化剂和导电基底的紧密结合,从而保证在长时间稳定工作;③易于调控原位生长的催化剂的形貌,从而有效地修饰电极的亲疏水性。为此,研究制备自支撑催化电极更能够满足商业化需求。
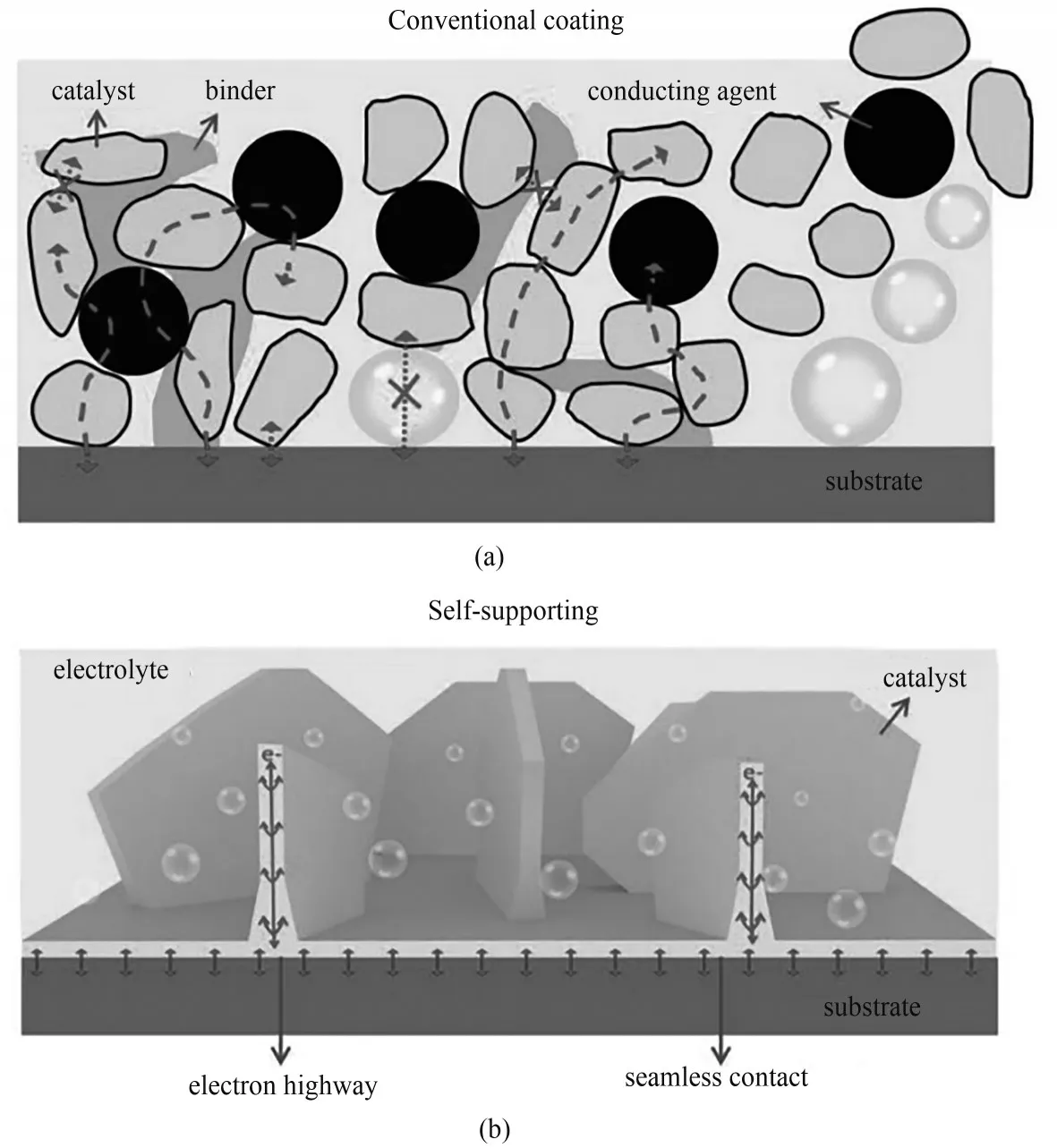
图2 两种不同的催化电极结构:(a)粉末状催化电极;(b)自支撑催化电极[9]Fig.2 Two types of electrocatalyst structure:(a)Catalyst in power form;(b)Self-supported catalyst[9]
为了发展自支撑析氢和析氧催化电极,不同的制备方法被不断开发和应用,不同的催化活性物质也被广泛研究制备。常见的自支撑催化剂制备方法有水热法/溶剂热法、电化学沉积法、化学气相沉积法、化学溶液法以及通过上述方法组合制备。如表2所示,具体总结了不同催化剂制备方法的优缺点以及适用场合。通常地,水热法/溶剂热法被认为是一种高效制备无机材料如金属氧化物、氢氧化物、羟基氧化物的方法,但需要在密压容器中反应。电化学沉积法是一种简单、高效且可操控性强的制备手段,通过调控沉积时间、电流密度或电压大小即可对催化剂负载量、形貌等进行调控。化学气相沉积法通常与水热法或电化学沉积法相结合,通过在对应温度下发生磷化/硫化反应,得到具有催化活性的磷/硫化物。而化学溶液法具有操作简单、反应条件温和、制备成本低的特点,在大规模制备用于工业化生产十分有优势。为此,本节主要围绕不同的制备方法制备①自支撑析氢催化电极;②自支撑析氧催化电极;③自支撑双功能催化电极。

表2 自支撑催化电极制备方法比较Table2 Comparisons of preparation methods of self-supported catalysts
2.1 自支撑析氢催化电极
近些年来,大量的研究工作集中在研究不同的高活性的析氢催化电极,如过渡金属硫化物、过渡金属磷化物、过渡金属碳化物、合金等。同样的,不同的基底也被广泛研究作为自支撑催化电极的基底,常见的主要有泡沫金属、碳布、碳纸、金属片等。
水热法是一种常见的制备自支撑析氢催化电极的方法。在水热过程中,基底表面的缺陷和含氧官能团可以作为成核活性位点,并限域表面活性物质的生长,从而使得活性物质在基底表面均匀生长。Miao等[11]通过一步水热法在碳布表面制备得到三维互连的Ni-Mo-S纳米片(Ni-Mo-S/C),他们以碳布作为基底,将其放入钼酸钠、L-半胱氨酸的混合溶液中,而后在200℃下反应即可以得到Ni-Mo-S纳米片。研究表明制备得到的Ni-Mo-S/C纳米片表现出了优异的析氢催化性能,仅仅需要200mV的过电位就能达到电流密度10mA·cm-2,同时,制备得到的催化电极同样拥有高稳定性,远远优于商业化的Pt电极。相似地,泡沫镍负载Ni2P纳米棒[12]、泡沫钴负载Co3Se4[13]、碳布负载二硫化钼[14]同样通过一步水热法制备,并表现出优越的催化析氢性能。
电沉积方法是另一种通用的制备自支撑催化电极的方法。电沉积过程是在电场作用下,将目标物质均匀地沉积在基底上。电沉积方法具有反应时间短、易于调控等优势,可以通过改变电沉积时间、电流密度或电压大小来调控催化剂负载量甚至改变催化剂的形貌,为此被广泛地使用。常见的物质,如过渡金属磷化物、过渡金属硫化物、氢氧化物等,都可以通过电沉积的方法制备得到。Guo等[15]以硫酸钴、次磷酸钠作为金属源和磷源,并加入柠檬酸钠当作络合剂,将碳纸作为工作电极,而后在恒定的电压下电沉积300~1800s,即可以在碳纸上沉积Co-P(Co-P/CP)。研究表明,当电沉积时间为600s时,碳布不能完全被Cu-Co-P微米球包覆,当增加电沉积的时间,可以发现Co-P在碳纸上的覆盖率明显提升;当电沉积时间达到900s时,可以发现碳纤维表面均匀覆盖一层Co-P微米球;当进一步增加电沉积时间,可以发现在均匀的Co-P微米球层上会进一步生长Co-P微米球。研究表面Co-P的催化析氢性能会随着电沉积的时间增加而增加,主要归因于随着电沉积时间的增加,催化电极的电化学双电层也增加。此外,泡沫铜负载的Ni-Mo电极[16]、泡沫镍负载的Co/CoP催化电极[17]也被制备作为高性能、高稳定的催化析氢电极。
此外,化学气相沉积法也被广泛运用于制备自支撑催化电极。在高温下,金属基底或者基底负载金属氧化物/氢氧化物/羟基氧化物在特定的气体氛围中转换为对应的化合物,如在PH3氛围中形成金属磷化物,在硫氛围中形成金属硫化物,在硒氛围中形成金属硒化物,在氨气氛围中形成金属氮化物等。例如,泡沫镍负载磷化镍纳米线的催化电极可以通过使用次磷酸钠作为磷源,泡沫镍作为金属源,然后在氩气氛围中300°C下反应2h得到[18]。在加热过程中,次磷酸钠受热分解为PH3,而后进一步与泡沫镍反应形成磷化镍。
另外,通过组合不同的制备方法也可以得到高性能的自支撑催化电极。如电沉积方法结合化学气相沉积法。如图3所示,Sun等[19]首先通过电化学沉积法在泡沫镍表面电沉积钴前体,而后在450℃将其转化为硫化钴,接着浸泡在金属镍溶液中,最后在450℃下磷化得到磷化镍修饰磷硫化钴的催化电极(CoS|Ni|P/Ni)。电沉积方法结合水热法。本课题组研究报道了一种蘑菇状结构的催化电极[20]。首先通过电沉积方法将氢氧化钴负载在碳布上,而后通过一步水热法将二硫化钼负载在钴前体上,同时氢氧化钴也转化为二硫化钴,即得到二硫化钴/二硫化钼复合电极,研究表明制备两组分间的作用显著地提升了电极的催化活性。电沉积法结合化学溶液法。本课题组[21]研究报道的具有核壳结构的Cu@CoP催化电极,通过该组合方法制备,在碱性和酸性条件下都表现出了优异的催化性能。
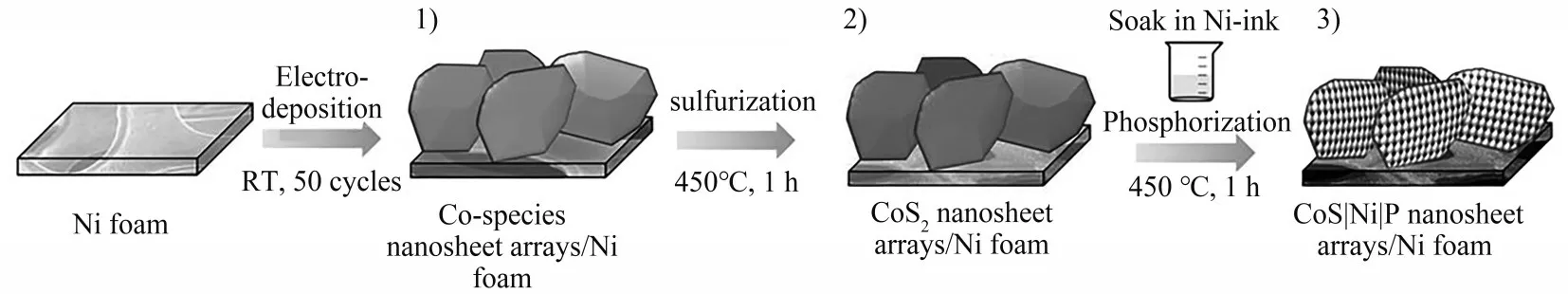
图3 制备CoS|Ni|P/Ni的示意图[19]Fig.3 Schematic diagram of preparation of CoS|Ni|P/Ni[19]
为了制备得到高性能的催化电极,除了以上讨论的水热法、电沉积方法、化学气相沉积法以及化学溶液法,目前还有许多其他制备方法被研究开发,如等真空抽滤法、等离子处理等。但是,这些都存在着制备工艺复杂、成本昂贵、过程烦琐精细、不易放大等问题。为了制备满足商业化可用的催化电极,以下三个方面需要进一步仔细考虑:①为了满足商业化大规模生产,发展能在常压常温下制备高性能催化电极的低能耗、简单安全的制备方法是最需要考虑的;②为了降低制备成本、提高制备效率,研究发展反应时间短、反应步骤少、制备步骤简单的制备方法同样值得关注;③催化电极的制备过程中,基底对催化电极的整体性能的贡献有待后续研究进一步明确,进而为制备高性能催化电极提供指导意见。
2.2 自支撑析氧催化电极
相比于析氢反应,析氧反应涉及一个四电子的反应途径,因而表现出了更强的动力学惰性。从而研究高效的析氧催化电极将可以显著地降低电解水的整体能耗。在酸性体系中,二氧化铱等贵金属催化剂被认为是具有高析氧活性的催化剂。但是,在碱性体系中,过渡金属衍生物表现更高的催化析氧活性,例如过渡金属氢氧化物、过渡金属氧化物、过渡金属羟基氧化物。因此,近些年来在自支撑析氧催化电极中,这些过渡金属基催化电极得到了广泛研究。同样的,类似于析氢催化电极的制备方法,上述制备方法同样适用于析氧催化电极。
化学溶液法是制备析氧催化电极的常见方法,通过将催化电极浸泡在指定的反应溶液中反应进而生成所需的催化活性物质。本课题组[22]报道了一种酸刻蚀方法制备得到高性能层状双金属氢氧化物(LOH)析氧催化电极,即NiFe LDH@NiFe的析氧催化电极。通过将泡沫镍铁浸泡在硫酸溶液中一段时间,而后干燥即得到NiFe LDH@NiFe。研究表明,这种制备方式能够实现催化基底和催化活性物质在原子层次上的紧密结合。制备得到的催化电极因此具有更高的电荷转移能力和催化稳定性,能够满足商业化应用的需求。相似的,Liu等[23]通过将泡沫铁浸泡在氯化镍和氯化钠的混合溶液中制备得到了高性能的NiFe(OH)x催化电极。Shao等[24]通过将泡沫铁浸泡在双氧水和硝酸钴的混合溶液中即可以制备得到Co-Fe羟基氧化物纳米片电极。由此可见化学溶液法是一种工艺简单、制备成本低、反应时间短的制备催化电极的方法。
化学气相沉积法也被广泛用于制备析氧催化电极。Yuan等[25]报道的NiSe2/NiFe2Se4@NiFe高效析氧催化电极,通过一步化学气相沉积反应,使用泡沫镍铁作为基底并同时作为镍铁源,使用硒粉作用硒源,在真空氛围下反应制得。析氧催化电极还可通过电化学沉积方法制备。Lu等[26]使用泡沫镍作为工作电极,使用硝酸镍、硝酸铁作为电解液,而后在恒压下进行电沉积,将非晶态的NiFe LDH沉积到泡沫镍表面。水热法同样适用于制备自支撑析氧电极,Zhang等[27]采用同源生长法,通过水热法将Ni(OH)2原位生长在了泡沫镍上,该制备方法可以实现催化剂和基底间的快速电子转移,并确保制备催化电极的高稳定性。
类似地,通过结合不同制备方法也是一种制备高性能析氧催化电极的途径。例如,通过化学溶液法与电化学掺杂法组合,本课题组[28]制备了一种铁掺杂氢氧化镍(Fe-Ni-OH)催化电极,首先将活化的泡沫镍浸泡在过硫酸铵溶液中,从而在泡沫镍表面原位形成氢氧化镍,而后将得到的中间体作为工作电极在硫酸亚铁铵溶液中电化学氧化,将可以实现铁掺杂在氢氧化镍中,从而使得制备得到的催化电极的催化性能得以显著提升。研究表明制备得到的催化电极在具有高活性的同时具有高稳定性。除了不同制备方法互相组合,还可以对同一制备方法多步耦合。本课题组[29]提出了一种多级结构的析氧催化电极,可以通过两步电化学沉积法实现。首先在大电流密度下将层级多孔镍沉积在镍网表面,进而在多孔镍表面沉积NiFe LDH,即得到多级结构的NiFe/Ni/Ni,该电极表现出了优异的催化性能。由此可知,通过组合制备方式可以实现多级结构催化电极的制备,是制备高性能催化电极的有效方法之一。
最后,对于析氧催化电极,由于其要在强氧化性环境下进行长时间的工作,为此对于自支撑催化电极来说,首先要确保其催化基底具有高耐碱性和强抗氧化性。但是,传统的自支撑催化基底,如碳布、铜基底、铁基底等在较高温下的抗氧化性较差,为此不能作为很好的自支撑催化基底。另外,催化基底对于析氧活性和稳定性的影响也需要进一步探讨。例如,Cano等[30]报道当泡沫镍作为析氧/氧还原反应的基底时,在长时间工作后,泡沫镍会部分溶解而后在催化剂表面形成羟基氧化镍/氢氧化镍薄膜,该薄膜会使得电极的传质能力下降。
2.3 自支撑双功能催化电极
通常来说,非贵金属催化剂如硫化物、磷化物、氮化物等具有高析氢催化性能。但是,非贵金属催化剂如过渡金属氢氧化物、过渡金属氧化物、羟基氧化物等则具有高析氧催化性能。而阳极和阴极使用不同的催化电极,这将会导致增加投资成本,同时不可避免地带来催化活性物质的交叉效应,使得电极催化活性显著地下降。为此,大量的研究工作集中在发展高性能的双功能催化电极,即耦合了具有高催化析氢活性催化物质与具有高催化析氧活性的催化剂。
为了实现不同催化活性物质的耦合,不同的制备方法被研究报道。例如,Ni3S2表现出了高析氧催化活性,MoS2表现出了高析氢催化活性,通过对这两种物质的结构设计,将可以实现高性能的双功能催化电极的制备。如图4所示,Yang等[31]通过两步水热法实现了MoS2-Ni3S2纳米棒催化电极的制备。首先通过一步水热法将钼酸镍纳米棒原位生长在泡沫镍表面,而后对中间体又通过一步水热法,实现了MoS2修饰Ni3S2纳米棒的制备,研究表明制备得到的MoS2-Ni3S2不仅存在大量异质结同时使得电子可以沿着Ni3S2快速传递,从而显著提升了HER和OER性能。另外,Zhang等[32]报道一种通过一步溶剂热法制备MoS2/Ni3S2催化电极的制备方法。通过将泡沫镍放置在硫代钼酸铵的二甲基甲酰胺溶液在200℃反应12h制备得到,研究表明MoS2/Ni3S2异质结可以促进含氧和含氢中间体的化学吸附,从而显著提升析氢和析氧的反应速率。为此,制备得到的MoS2/Ni3S2催化电极具有高双功能催化性能。另外,Cao等[33]也报道了通过一步水热法制备MoS2/Ni3S2纳米阵列的制备方法,通过将泡沫镍浸泡在硫脲和钼酸钠的水溶液中在180℃反应得到,制备得到的催化电极同样表现出了高析氢、析氧催化活性。

图4 MoS2-Ni3S2纳米棒制备示意图[31]Fig.4 Schematic illustration for the synthesis of MoS2-Ni3S2nanorod[31]
另外,也可以通过简单的步骤组合催化活性物质来制备双功能催化电极。例如NiCoP具有高析氢活性,NiFe LDH具有高析氧活性,Zhang等[34]首先通过水热法和化学气相沉积法在泡沫镍表面原位生长NiCoP纳米线,而后又通过一步水热法将NiFe LDH纳米片原位生长在NiCoP纳米线上(NiFe LDH@NiCoP/NF),实现了高性能双功能催化电极的制备。研究表明两组分之间的协同作用能够显著地提高结构稳定性和催化析氢、析氧性能,将制备得到的NiFe LDH@NiCoP/NF催化电极作为析氢和析氧催化电极,仅仅需要1.57 V的电压就能达到10mA·cm-2。相似地,Yang等[35]在泡沫镍表面上成功合成一种核壳结构Mo-Ni2P@NiFe LDH催化电极,该特殊结构使得催化电极的传质能力和电子转移能力都显著增强,从而具有高析氢、析氧催化性能。另外,Mai等[36]首先结合水热法和化学气相沉积法制备得到CoxMoP中间体,而后又通过一步电沉积法将NiFe LDH原位沉积到CoxMoP表面,得到CoxMoP/NiFe-LDH催化电极,研究表明两组分之间的强电子作用和大量暴露的活性位点是CoxMoP/NiFe-LDH电极具有高催化活性的主要原因。由此可见,使用组合制备方法制备催化电极具有工艺简单、结构易于调控等优点。但是,通常来讲,两组分之间的耦合在某些情况下由于其传质和暴露的活性面积受到了限制,反而会抑制单组分的活性,在设计双功能催化电极时,需要全面考虑。
研究自支撑催化电极从电极的稳定性、催化活性、工艺制备等各个指标来说都具有重要的意义。未来进一步研究开发具有制备方法简单、常压下制备、反应时间短等特点的制备方法将能加速自支撑催化电极的商业化,推动电解水技术的发展。
3 耐碱腐蚀的离子膜
3.1 离子膜技术要求
电化学能源转化与储能过程对膜材料提出新要求,特别是材料耐电化学腐蚀、孔径分布和高度稳定性方面,远远超过作为“分离介质”的膜材料性能。通常来看,用于电能转化与储能的膜材料性能方面,需要满足以下几方面要求。
(1)离子传导性。在电化学装置运行过程,需要通过溶液中的离子定向迁移,才能形成电池内电流,要求电池隔膜具备良好的传导离子特性。通常使用膜面电阻来表征,膜面电阻的值越小,表明其传导离子的能力越强。
(2)高选择性。电化学装置中的隔膜起着分隔氧化剂与还原剂的作用,为了避免氧化剂和还原剂接触发生反应,降低能量效率,要求膜材料必须有效阻隔电化学活性物质透过。膜的选择性直接影响装置的库仑效率。例如,在质子交换膜燃料电池(PEMFC)中的隔膜,需要阻止氢气渗透并导通氢离子;全钒液流电池(VFB)中的隔膜,需要阻止不同价态钒离子渗透,使氢离子快速通过。
(3)高稳定性。电池过程包含氧化反应半电池和还原反应半电池。在氧化反应半电池中存在强烈的夺取电子趋势,离子传导膜长期工作在强氧化性环境中,要耐受新生态氧等物质的腐蚀,对膜材料的稳定性提出十分苛刻的要求。
除此以外,膜材料制备过程是否容易工程放大,制膜原料易得性,制膜工艺稳定性、制膜过程连续性,这些因素与膜产品质量可靠性、成本合理性密切相关,直接决定是否能够满足新兴能源市场的迫切需求。
3.2 电解水用阴离子交换膜
尽管PEMWE过程在一定条件下具有独特的优势,但由于其特定的酸性环境,阴阳两侧的析氢和析氧催化剂选择十分受限,主要由Pt系贵金属催化剂组成。为解决贵金属资源在大规模制氢应用中受限的问题,以及降低材料成本,阴离子交换膜电解水(anion exchange membrane water electrolysis,AEMWE)在2012年应运而生。通过将传递H+的质子交换膜更换为传递OH-的阴离子交换膜(或称碱性膜),析氢和析氧反应都得以在碱性环境中发生,因此诸如Ni、Fe等非金属催化剂都能够稳定地应用于催化过程,这也使得AEMWE具有广泛的应用前景[37]。
在AEMWE过程中,阴离子交换膜承担着重要作用。OH-作为一种生成物、反应物和载流体,借助阴离子交换膜能够从阴极侧传递至阳极侧。同时阴离子交换膜也起到阻隔气体和分隔两极的作用。但与发展较为成熟的质子交换膜不同,商业化阴离子交换膜大多受限于其较差的耐碱稳定性及较低的OH-传导率[38]。由于该材料在制氢领域的应用前景和现存的问题,近年来受到广泛关注。
一般来说,阴离子交换膜分子结构上主要由高分子骨架和阳离子基团构成。高分子骨架作支撑材料提供力学性能,而阳离子基团则与水共同作用帮助OH-在电场作用下进行定向迁移。发展较早的一系列阴离子交换膜其主要思路是通过对现有的工程塑料进行接枝改性,包括聚苯醚、聚醚砜、聚醚醚酮等。然而此类聚合物主链均存在醚键,在高温、强碱性的环境下,醚键受到OH-进攻容易发生断裂,导致分子量的降低及膜的破裂[39-40]。近期针对电解水阴离子交换膜的研究中,发展了大量从单体结构和聚合反应的角度进行设计的无醚键主链,极大程度地改善了这一类问题。基于超酸催化反应,Cha等[41]制备了主链无醚键的高性能阴离子交换膜,由于化学结构的稳定性,该材料能够在热碱中实现超过1000h的电导率维持,且在1.9 V的电解电压下实现3.5 A·cm-2的制氢电流。Soni等[42]也通过钯催化偶联反应得到了高分子量的全芳香主链阴离子交换膜,实现了在纯水/碱液中超100h的稳定电解,且纯水电解电流达到500mA·cm-2。
阳离子基团方面,较早的研究中主要以三甲基季铵盐为功能基团,但研究表明,诸如此类的季铵盐在高温碱性的环境下,容易被OH-亲核进攻而发生取代或β消除反应,进而使阴离子交换膜降解而失去传递OH-的能力。针对这一点,近年来的研究工作也对稳定阳离子基团的结构作出了大量理论设计和验证,并开发出包括芳香类季铵盐、非芳香环胺型盐、季鏻盐、金属中心阳离子等多种化学结构稳定的功能基团。Fan等[43]针对咪唑这一芳香型季铵盐的耐碱性进行探究,通过理论预测和实验筛选了耐碱半衰期超过10000h的咪唑盐结构,并基于Yamamoto偶联反应制备了能够在6mol·L-1碱液中稳定电解水的阴离子交换膜。
为了从阴离子交换膜材料中有限的阳离子基团密度上提升膜整体的OH-传导能力,需要将膜内的离子基团进行更加有效的定向排列,以形成一定的离子通道。通过引入和调节多种分子间的作用力,能够促进功能基团自发组装和排列,并构成聚合物内部的微相分离,从而获得OH-传导率更高的阴离子交换膜材料。基于两种工程塑料聚苯并咪唑和聚氯甲基苯乙烯的交联反应,可以极为简单地制备具有良好微相分离效果的阴离子交换膜。该类由两种不同主链共同形成的离子通道表现出优异的OH-传导能力,在AEMWE的应用中能够在2.0 V电解电压下实现700mA·cm-2的电流密度[44]。Li等[45]则通过实验证明了保证一定程度IEC对AEMWE过程中形成离子通道,强化OH-传递的重要性。通过采用IEC高达2.6mmol·g-1的聚合物阴离子交换膜HTMADAPP配合高IEC的碱性聚合物binder,能够实现1.8 V下超过1600A·cm-2的纯水电解电流。
3.3 溶剂化的离子膜电解水
虽然前文所述的PEMWE过程电解水能够实现较高的产氢速率,而AEMWE过程也能够在保证一定的电解效率的同时,避免贵金属催化剂的使用。然而,上述两个膜法电解水过程都无法避免对离子交换膜材料的应用。离子交换膜材料是电解水组件中占据大量成本的一种关键材料,也是影响电堆持久运行的弱点材料,其中质子交换膜如Nafion膜成本高,阴离子交换膜稳定性差,难以制备,在引起广泛研究者关注的同时,也制约着膜法电解水技术的发展[46]。
为解决上述问题,一些研究者采用非离子交换膜代替离子交换膜作为电解水的膜材料,并且取得了令人满意的成果。由于类似的方法所选用的膜材料亲水性好,能够与溶剂分子结合,因此将该类膜材料统称为溶剂化膜。例如,Shi等[47]使用反渗透膜进行海水电解制氢。由于反渗透膜的售价是离子交换膜的十分之一,因此该过程的经济性被极大程度地提高了。此外,反渗透膜有较好的离子选择性,能够使H+和OH-快速通过的同时,截留较大的其他离子,实现高选择性的制氢过程。类似地,聚苯并咪唑材料由于其独特的化学结构,能够与碱进行掺杂成为溶剂化膜,且该状态下具有高离子传导优势。研究证明,通过亲水聚合物掺杂能够进一步凸显该优势,在1.9 V下能够达到0.1 A·cm-2的电解电流[48]。
由于大多数聚合物膜材料在强酸性或碱性的电化学体系下都难以保证绝对的稳定性,报道的电解水膜材料在实际使用过程中都不可避免地会随时间存在不同程度的降解。然而,本课题组[49]开发的无机纳米粒子复合型的溶剂化膜则能够保持近乎无衰减的电解水过程。如图5所示,该材料PTFE/LDH采用高稳定的聚四氟乙烯作为基材,通过原位生长的方式将Ni/Fe层状氢氧化物进行孔填充,作为溶剂化粒子。由于该类材料是在碱性环境中制备的,因此不存在降解途径。实验表明,PTFE/LDH能够维持2000h电导率无变化,且在1.8 V下的电解电流达到1A·cm-2,其性能远超商业化的fumatech离子隔膜(FAA-3)。引人注意的是,该材料制备方法极其简单,成本较低,是一种具有工业化应用前景的电解水膜材料。
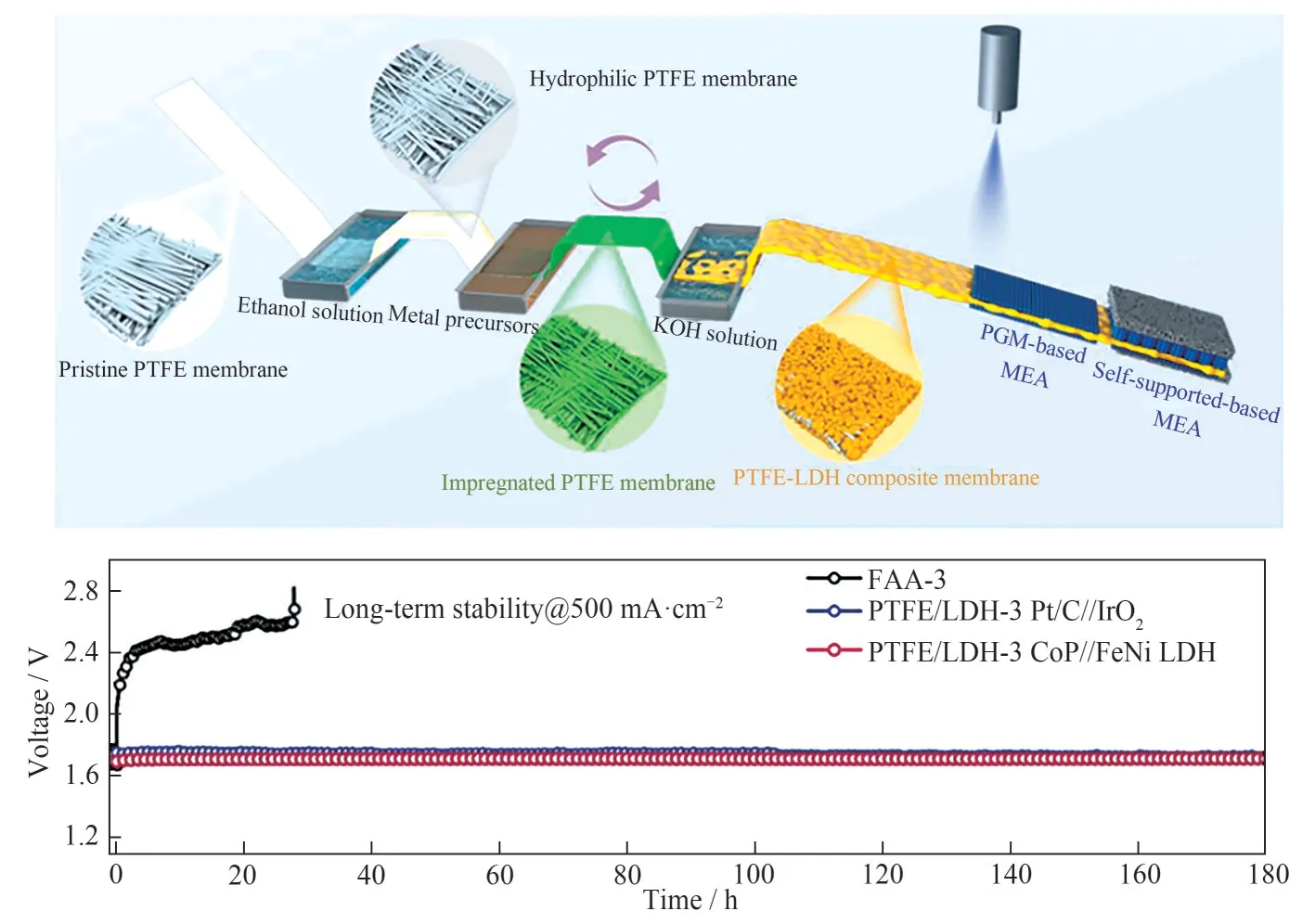
图5 PTFE/LDH无机纳米粒子复合填充膜制备及高稳定电解水测试[49]Fig.5 PTFE/LDH membrane preparation and water electrolysis performance[49]
3.4 酸性/碱性双性膜电解水
将电解水过程分为两个部分,应该由阴极析氢反应和阳极析氧反应两个电化学半反应构成。从动力学角度考量,析氢反应在酸性条件下过电位较低,而析氧反应则在碱性环境中有利。然而,无论电解水过程是在酸性还是在碱性中进行,都无法同时在两侧均保持动力学的优势[50]。
基于以上科学问题,本课题组[51]提出使用一张膜,将酸性和碱性溶液隔开,使得两侧电极均处在优势的pH溶液中,实现过电位的降低,进而减小电解水的能耗。由于酸碱两性的引入,电解水的平衡热力学电位差值从1.23 V降低至0.401 V,而电解实验也实现在1.23 V以下达到电解电流超过150mA·cm-2。最初,受限于能够稳定的将酸碱隔开的膜材料,该过程只能依靠Nafion阳离子膜完成,而实验中也确实观察到酸碱中和的现象。在该工作的基础上,将膜材料改为聚苯并咪唑系列[52],并借助该结构能够与酸碱溶剂化的独特优势,降低酸碱中和发生的速率,提升了该过程的稳定性。
Boettcher等[53]则通过双极膜的电解实验证明,双性电解水过程要对单性电解水形成竞争,双极膜中间层的水解离催化剂是关键因素。并且,该层催化剂还应随着位置不同提供不一样的酸碱性环境。基于以上认知,双极膜电解水实现了800mA·cm-2的电解性能,且体现出较低电流下的优势。在此基础上,Mayerhöfer等[54]进一步验证了,双极膜中间层的三维结构设计能够在同样的推动力下为电解水过程提供更高的电解速率。
4 结构有序的膜电极
传统的碱性水溶液电解基于液态碱性电解液和多孔膜。为了防止产生的气体交叉渗透,碱性溶液电解的电极间隙一般在2mm以上。这对电解槽的安全高效运行至关重要。由于电解液和电极表面黏附气泡的高欧姆内阻,碱性溶液电解的电流密度较低(250mA·cm-2),能量转化效率通常只有60%。特别是气泡的产生对电解性能有显著的负面影响[55]。为了追求高性能的碱性电解槽,通过借鉴质子交换膜电解槽的结构,使用阴离子交换膜取代多孔隔膜,发展了零间隙碱性电解槽[56]。通过减少阴离子交换膜两侧的两个多孔电极来设计零间隙型电解槽,从而使电极之间的电压降最小。
膜电极组件(membrane electrode assembly,MEA)作为零间隙型电解槽的核心部件,是电解水过程中进行电化学反应和传质的场所,是决定电解性能和稳定性的重要因素[57]。MEA是一种由多孔气/液扩散 层(liquid-gas diffusion layer,LGDL)、催 化 剂 层(catalyst layer,CL)和膜组成的夹层结构。以AEM电解为例,水通过电催化剂与电子发生电化学反应,并裂解成氢离子和氢氧根离子。然后,氢氧根离子通过AEM传输,在阳极侧形成氧气。同时,氧气和水在阳极侧被输送出去。值得注意的是,碱性离聚物的利用提供了碱性环境,导致电解液是纯水而不是碱性溶液。这降低了电解槽部件腐蚀的可能性。在电解过程中,电化学反应只发生在“三相边界”(three-phase boundaries,TPBs)上,即有电子导体、活性催化剂、氢氧化物离子载体和反应物/产物路径的位置[58]。例如,阳极的水分解位置需要①一个孔来输送液态水和气态氧气,②一个电催化剂和电子导体来进行反应,③一个电解质来输送氢氧根离子。
MEA的传统制备工艺根据催化层的支撑体不同可分为:CCM(catalyst-coated membrane)法和CCS(catalyst-coated substrate)法 两 种[37]。CCM法 以 干 燥的聚合物膜为支撑体,将催化层以多种方式结合于聚合物膜两侧形成MEA,该方法制备的MEA具有三层结构,将以该工艺制备的MEA记为CCMMEA;CCS法以气体扩散层为支撑体,首先将催化层以多种方式依附于气体扩散层上制备多孔气体分散电极,最后将多孔气体分散电极与聚合物膜通过热压方式结合成MEA,该方法制备的MEA具有五层结构,将以该工艺制备的MEA记为CCS-MEA。
从功能角度来看,膜电极实现了催化反应与传质二者功能一体化,既能够实现隔离氧化剂、还原剂功能,又能够在相对温和条件下完成大规模电能转化与储能,以及制造特定化学品。为了进一步提升膜电极性能,通过开发表面图案化技术,设计和优化离子、电子的传质界面,有望进一步降低电能转化与储能过程的能量损耗。
4.1 CCS型膜电极
对于CCS型膜电极的制备过程,通常在LGDL(金属泡沫或金属网状基底)上通过多种方法涂覆催化剂,形成气体扩散电极,紧接着与膜热压形成膜电极结构。该种膜电极制备方法具有工艺成熟、成本低廉和操作简便等优点[15,59]。在传统的气体扩散电极制备过程中,首先对导电基底进行预处理,然后将配制好的催化剂浆料与LGDL结合制备气体扩散电极[60]。因此,传统气体扩散电极中的催化层是由粉末催化剂和黏结剂(Nafion或PTFE等)组成的导通网络。该催化层结构具有以下劣势:①使用聚合黏合剂,不可避免地增加了传质阻力,并覆盖了催化活性位点。②粉末催化剂与导电基底间的黏附力较低,导致粉末催化剂的负载量通常小于1mg·cm-2,提供的催化活性位点有限。③在长时间或大电流密度电解过程中,粉末催化剂容易从导电基底上脱落。而自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[61]。因此,近几年围绕自支撑电极的设计与制备取得显著进展。
Kim等[62]报道了一种简易的策略制备PEM电解用Ir基气体扩散电极。通过电化学和热化学氧化法,成功在碳纸基底上原位制备出高本征活性的IrOx催化层,并且气体扩散电极的Ir载量可低至53.9 μg·cm-2。利用Ir基气体扩散电极作为PEM电解的阳极,电池电压为1.6 V时的质量比电流密度高达6.8 A·mg-1。
Lee等[63]通过一步原位刻蚀的方法成功在泡沫镍基底上制备得到高性能的OER催化层。利用Fe3+对Ni基底进行刻蚀,在泡沫镍表面形成堆积致密的Ni0.75Fe2.25 O4纳米颗粒。在1mol·L-1KOH溶液中,该电极在10mA·cm-2电流密度下的OER过电势仅为192mV,比商业化的IrO2贵金属催化剂低73mV。当与Pt/C HER催化剂结合时,在1mol·L-1KOH中,电池电压为1.9 V时,AEM电解的电流密度高达2.0 A·cm-2。在500mA·cm-2电流密度下进行电解稳定性测试,AEM电解的能量转化效率始终维持75%以上。
除了使用电化学和化学溶液法等方法在导电基底上原位构建催化层,溶剂热、离子溅射和化学气相沉积等方法同样被广泛用于自支撑电极的制备[64-66]。以气体扩散层为催化层支撑体的制备方式中,在控制催化剂组成、结构及载量等方面具有一定优势,但是存在催化层与聚合物膜的结合较差问题。
4.2 CCM型膜电极
为了提高CCS-MEA中催化层与聚合物膜的界面结合力,研究人员开发出了CCM型MEA[67]。该制备工艺特征是以聚合物膜为催化层的支撑体。在制备过程中,首先需要制备具有一定机械强度的聚合物膜,并进行预处理。同时配制催化剂浆料:纳米催化剂、异丙醇、黏结剂等一定比例超声混合均匀。然后通过不同的方式将催化剂浆料与聚合物膜结合,进而在聚合物膜表面形成催化层。待烘干后,最后与气体扩散层组装。在CCM制备工艺中,催化层的形成将显著影响MEA的性能,因此催化剂浆料涂覆于聚合物膜表面的方式就显得格外重要。
Feng等[68]利用贴纸转印法成功制备了CCMMEA。如图6所示,催化剂浆料首先被喷涂在薄且平坦的PTFE板(0.1mm)。Nafion膜被夹在两个催化剂涂层的PTFE薄片间,通过在30kg·cm-2和135℃条件下热压3min,催化层被转印到活性面积为2cm2的聚合物膜两侧。得到的CCM被放置于两张碳纸气体扩散层间形成CCM-MEA。将该CCMMEA用于PEM电解,在25℃条件下1.8 V电压处的电流密度高达600mA·cm-2。
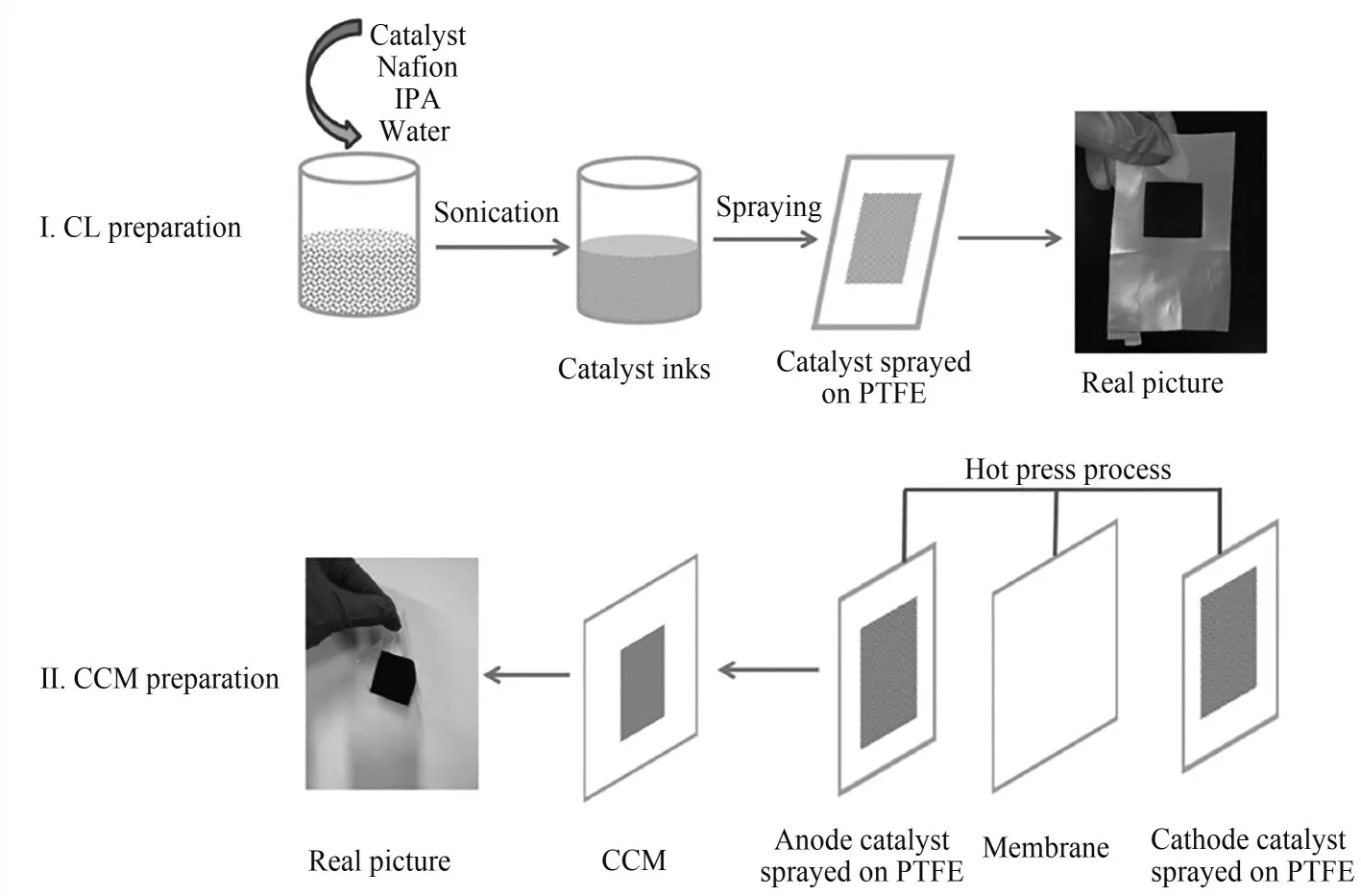
图6 基于贴纸转印法制备CCM-MEA的流程示意图[68]Fig.6 Preparation of CCM-MEA based on transfer methods[68]
为了进一步提高催化层与聚合物膜间的界面强度,Wang等[69]通过在120℃下加热非晶态Nafion膜和催化层,成功制备了一种共晶型CCM-MEA,并用于PEM电解。该共结晶处理有效地加强了聚合物膜与催化层之间的键合,并提高了催化层的疏水性。因此,与传统CCM-MEA相比,采用共晶型CCM-MEA的PEM电解稳定性显著提高。
综上所述,以聚合物膜为催化层支撑体的制备方式中,除了转印和涂覆这两大类将催化层与聚合物膜结合的方法,还包括电化学沉积等多种方法。总之,这些方法始终围绕着如何增强催化层与聚合物膜的结合力、提高催化剂利用率、简化操作工艺等方面进行改进。
4.3 结构有序的膜电极
除了CCS-MEA和CCM-MEA两种类型膜电极,另外一种是构建具有合理设计的催化剂层的MEAs[70-71]。Zeng等[72]提出了一种理想的PEM燃料电池催化剂层结构,其催化剂层厚度超薄,孔隙垂直排列,活性位点位于电子和离子导体的边界;这种结构有利于反应物、电子和质子的传输。数学模型预测和实验研究表明:有序结构的MEA比传统的MEA电池性能有很大提高。该类有序化MEA被普遍认为是第三代MEA。同样地,近年来诸多研究表明:在电解水系统中,与传统结构的MEA相比,有序化MEA具有提高催化剂利用率、降低成本及增强气液传质等优势[73-75]。
传统的CCM-MEA在目前商业化的PEM电解中的平均Ir贵金属负载量为2~3mg·cm-2,导致PEM电解制氢成本高昂,难以大规模应用[76]。为降低贵金属Ir的负载量并且保持高效的催化性能,Jiang等[77]设计并制备了一种缺陷Ir薄膜包覆的WOx纳米棒的有序化电极(图7)。在有序化的WOx支撑体上均匀分散的Ir催化剂有利于提高Ir质量比活性,并提高WOx的导电性。因此,将低Ir负载量(0.1 4mg·cm-2)的有序化MEA用于PEM电解,在80℃条件下,2.0 V电压处的电流密度高达2.2 A·cm-2,并且以500mA·cm-2电流密度稳定电解1030h。
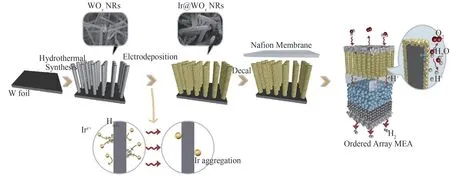
图7 Ir@WOx纳米阵列有序化膜电极的制备与结构示意图[77]Fig.7 Preparation of Ir@WOx nano array as aligned MEA[77]
为了增强AEM电解过程中的气液传质过程,Chi等[78]报道了一种简单的水热合成方法,以泡沫镍为基底,成功制备了基于无黏结剂、垂直排列的FeOOH/NiFe层状双氢氧化物(LDHs)纳米片的有序化MEA,并用于AEM电解。无黏结剂和垂直排列的纳米结构可以显著提高电化学表面积,有助于在纳米尺度上形成有效的化学/界面分布和快速的离子-电子转移。另外,纳米片之间的间隙缓解了氧气析出过程中催化层的剥离,有利于建立更稳定的TPBs。因此,将该有序化MEA用于AEM电解,在1.71 V电压处实现200mA·cm-2的电流密度,对应高达86%的能量转化效率,显著优于商业化碱性电解水。
膜电极作为电解水过程的核心部件之一,提高其性能和寿命、降低其成本具有非常重要的意义,可以加速聚合物膜电解水大规模商业化进程。MEA的发展经历了从第一代CCS型到第二代CCM型,再到有序化MEA的发展阶段。开发制备工艺更简单、性能更好更稳定、成本更低的有序化MEA,促进有序化MEA在聚合物电解水中的商业化应用是未来MEA研究的主要方向。
5 结 语
由于风能、太阳能等可再生能源技术发展,以及燃料电池为动力的交通能源需求,氢气作为清洁高效的能源载体,引起国内外高度关注。高效率电解水制氢技术,将成为未来新能源产业的战略制高点。通过比较现有的电解水制氢技术路线,从大规模、长寿命、低成本的综合角度分析,认为碱性膜电解水制氢具备独特优势。一方面,与商业化的碱性水溶液电解水制氢相比,可以降低内阻、提高电流密度和能量效率;另一方面,可采用非贵金属制备催化剂,克服质子交换膜电解水制氢受限于贵金属催化剂,导致建设成本昂贵、资源受限问题。本文通过系统归纳自支撑催化电极、耐碱腐蚀离子膜、有序结构膜电极等关键材料研究进展,指明碱性膜电解水制氢装备与技术发展趋势。在此基础上,需要加强催化电极、离子膜等关键材料制备过程的机理研究,尤其是揭示电化学反应器的一般性规律。从功能角度来看,膜电极实现了催化反应与传质二者功能一体化,既能够隔离产生的氢气和氧气,又能够在相对温和条件下完成大规模电能转化与储能,满足低碳减排的产业需求。从催化剂制备策略到耐碱离子膜发展趋势,以及有序化膜电极的优势应用,解明电化学过程中的传质与反应耦合原理,将有力推动先进的电解水制氢装备与氢能产业技术进步。
