毕赤酵母新型反向筛选标记基因的鉴定
2022-01-05牛硕朱梅君魏子贡
牛硕,朱梅君,魏子贡
(湖北大学生命科学学院, 湖北 武汉 430062)
0 引言
甲醇营养型毕赤酵母(Pichiapastoris)作为目前生产重组蛋白最重要的表达系统之一,具有分子遗传简单、能够对表达的蛋白进行翻译后修饰等优点[1].随着分子生物学的发展,在基因水平对毕赤酵母进行基因改造成为研究毕赤酵母基因功能的主要手段.
对于酵母菌的遗传操作中,经常使用标记基因来筛选重组菌株,然而目前应用广泛的标记基因主要是编码抗生素的基因[2].对于毕赤酵母而言,能够被实际运用于基因工程的标记基因数量有限,而且进行多次基因操作时,由于可供选择的筛选标记会受到限制,所以在筛选经过多次基因修饰的重组菌株时就会相对困难.利用多个标记基因筛选重组菌株也会增加前期载体构建的复杂程度,因此有效地消除标记基因可以克服这些难题.
利用反向筛选标记实现无标记基因操作的方法为微生物基因工程研究提供了新的工具[3].在微生物领域中,目前常用的反向筛选标记包括大肠杆菌(Escherichiacoli)中编码半乳糖激酶的galK基因,galK基因的表达令E.coil菌株对2-脱氧半乳糖敏感,在2-脱氧半乳糖存在时,中间代谢2-脱氧半乳糖-1-磷酸会在细胞内积累,导致细胞死亡[4];E.coil中mazF基因编码毒素蛋白MazF,MazF是一种序列特异性核酸内切酶,作为mRNA干扰酶,识别mRNA的ACA序列,拆开单链mRNA,从而阻断蛋白质的合成,导致细胞生长受阻[5-6];酿酒酵母(Saccharomycescerevisiae)中的URA3基因编码乳清苷酸脱羧酶,该酶是合成尿嘧啶的关键酶,可催化5-FOA(5-氟乳清酸)转化为有毒物质,导致细胞死亡[7].以上的研究表明,无标记基因操作体系在分子生物学中的重要作用以及广泛应用.利用无标记基因操作的方法可以实现基因的敲入、敲除、定点突变等基因操作[8],筛选标记回收后还可以重复利用,从而对同一个宿主菌株进行多次基因修饰且不会引入多余的标记基因.
雷帕霉素(Rapamycin,RAPA),又名西罗莫司,属大环内酯类抗生素,为低毒性抗真菌药物.使用雷帕霉素处理酵母后,细胞周期不可逆转地停滞在G1期,阻断细胞周期由G1期至S期的进程[9].本研究通过参考Zhu等[10]利用转座子突变技术构建毕赤酵母突变文库的方法,建立了百万级别的突变库,通过筛选获得具有雷帕霉素抗性的毕赤酵母突变株mut375.通过热不对称交错PCR(Thermal asymmetric interlaced PCR,TAIL-PCR)实验确定了突变株的突变位点,转座子插入了PAS_Chr2-2_0375基因,导致该基因的功能缺失.由于毕赤酵母PAS_Chr2-2_0375基因表达产物与酿酒酵母的FPR1基因产物有高度同源性,因此将该基因命名为kFPR1.
实验表明kFPR1基因的缺失会导致毕赤酵母产生雷帕霉素抗性表型,把携带kFPR1基因片段的表达载体pGAPZB-kFPR1转化至KO菌株,发现重组菌株恢复了对雷帕霉素的敏感性.将突变株mut375与野生型毕赤酵母GS115菌株在YPD固体培养基中的生长情况进行对比,突变株与野生型酵母的生长情况相近,表明kFPR1基因的缺失并不会影响毕赤酵母的生长,这些结果显示kFPR1基因具有作为毕赤酵母反向筛选标记的特性.以EGFP(Enhanced Green Fluorescent Protein)作为目的蛋白,博来霉素抗性基因(ZeoR)和kFPR1分别作为正向筛选标记和反向筛选标记,成功将EGFP基因整合至毕赤酵母染色体上并且回收了筛选标记片段ZeoR-kfpr1,证明了kFPR1作为毕赤酵母反向筛选标记的可行性.
本研究利用转座子突变技术筛选得到具有雷帕霉素的突变体,并证明了毕赤酵母kFPR1基因的缺失会导致毕赤酵母产生雷帕霉素抗性表型,可以通过导入携带kFPR1基因的表达载体恢复对雷帕霉素的敏感性.因此,利用kFPR1基因作为反向筛选标记,建立了一种适用于毕赤酵母的无标记基因操作方法,为毕赤酵母的遗传操作提供了新的筛选工具.
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 本研究所用的毕赤酵母菌株见表1,毕赤酵母表达载体pGAPZB、pPICZA及大肠杆菌克隆载体pVAX1均购自Invitrogen公司,重组载体pPICZA-EGFP由实验室前期构建.

表1 本研究使用毕赤酵母菌株
1.1.2 培养基及试剂 大肠杆菌生长所用LB培养基含0.5%酵母粉,1%蛋白胨,1%氯化钠;毕赤酵母生长所用完全培养基(YPD)或选择性培养基(YND、YPDZ和YPDR).YPD培养基含1%酵母粉,2%蛋白胨,2%葡萄糖;YND培养基含0.17%酵母氮源基础(无氨基酸无硫酸铵),0.5%硫酸铵,1%葡萄糖;YPDZ培养基即YPD培养基中添加100 μg/mL 博来霉素;YPDR培养基即YPD培养基中添加100 ng/mL雷帕霉素;酵母裂解液含0.2 mol/L 醋酸锂,0.5 mol/L氯化钠,0.01 mol/L EDTA(pH8.0),0.1 mol/L Tris-HCl(pH 8. 0),1% SDS.
1.1.3 引物 本研究使用的引物见表2.

表2 本研究所用引物
1.2 方法
1.2.1 突变株mut375的筛选与分离 利用转座子突变技术构建毕赤酵母细胞GS115的突变体库[10],将辅助型质粒和供体型质粒转化至GS115感受态细胞,涂布至YND固体培养基,收集平板上的菌落,将获得的酵母菌液富集后形成突变库.取少量菌液分别涂布至YPDR固体培养基,30 ℃培养48 h,得到能够在YPDR培养基上生长的突变株,命名为mut375.
1.2.2 获取突变株mut375中转座子插入位点的侧翼序列 将具有雷帕霉素抗性的突变体单菌落在YPDR培养基中培养至OD600=0.8~1.0时,取1 mL菌液,使用酵母裂解法提取突变体基因组DNA[11],分别以LAD1-1/SBp1和AC1/SBp2为引物,通过热不对称交错聚合酶链式反应(TAIL-PCR)得到转座子插入位点侧翼序列的扩增产物,具体PCR程序参考Liu等[12]的研究.将PCR产物进行测序,从而获得mut375突变株转座子插入位点侧翼序列(见图1).
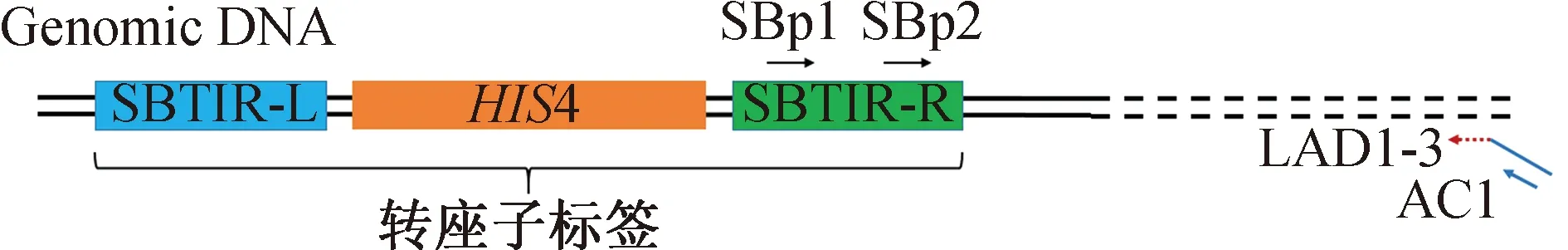
图1 获取转座子侧翼序列
1.2.3 转座子在染色体上的插入位点 利用生物信息学网站NCBI(national center for biotechnology information)在线BLAST工具(https://blast.ncbi.nlm.nih.gov/Blast.cgi/)将mut375突变株转座子插入位点侧翼序列与毕赤酵母GS115基因组文库进行比对,得到转座子在染色体上的插入位点.
1.2.4 pVAX1-HIS4-kfpr1HA载体的构建 为了敲除毕赤酵母的kFPR1基因,本研究构建了携带HIS4基因表达盒的同源臂片段.用EcoRI和XhoI消化质粒pVAX1,胶回收线性化片段;以毕赤酵母GS115基因组DNA为模板,使用kfpr1HA-UP-F/kfpr1HA-UP-R引物扩增得到kfpr1HA-Up片段,该片段为648 bp的kFPR1基因上游同源序列;使用kfpr1HA-DO-F/kfpr1HA-DO-R引物扩增得到kfpr1HA-DO片段,该片段为552 bp的kFPR1基因上游同源序列;以pPIC9K质粒为模板,使用His4-F/His4-R引物扩增得到His4-up/do片段.回收上述扩增得到DNA片段,参照CloneExpress® MulitS One Step Cloning试剂盒的说明书的操作步骤将这4个DNA片段无缝连接,将反应液转化至大肠杆菌TOP10感受态中,用添加了氨苄青霉素(100 μg/mL)的LB固体培养基筛选出阳性转化子,使用Plasmid Mini kit I(Omega)提取质粒,将质粒命名为pVAX1-HIS4-kfpr1HA(图2).
1.2.5kFPR1基因缺失的KO菌株 质粒pVAX1-HIS4-kfpr1HA用EcoRI和XhoI酶消化,释放HIS4- kfpr1HA片段,胶回收酶切产物,将HIS4-kfpr1HA片段转化至毕赤酵母GS115感受态细胞中,涂布至YND固体培养基.利用同源重组将该片段整合至kFPR1基因处,实现kFPR1基因的敲除.挑取单菌落进行PCR验证,测序正确的转化子即为kFPR1基因缺失的KO菌株.
1.2.6 pGAPZB-kfpr1载体的构建及功能回补的TR菌株 以GS115基因组DNA为模板,使用ExkFPR1-F/ExkFPR1-R引物扩增kfpr1片段,与EcoRI和XhoI双酶切处理pGAPZB质粒的回收产物无缝连接,得到pGAPZB-kfpr1载体.用AvrII消化pGAPZB-kfpr1质粒,得到线性化的pGAPZB-kfpr1载体,转化至KO菌株制备的感受态细胞,经过PCR实验验证后得到kFPR1基因功能回补的TR菌株.
1.2.7 构建pPICZA-EGFP-CYC1TT-kfpr1载体 以pPICZA质粒为模板,使用Cyctt-F/Cyctt-R引物扩增CYC1TT片段;以pGAPZB-kfpr1质粒为模板,使用pGAP-F/kfpr1-R引物扩增pGAP-kfpr1片段;用EcoRI和XbaI消化pPICZA-EGFP质粒的胶回收产物,3个DNA片段进行无缝连接,得到pPICZA-EGFP-CYC1TT-kfpr1质粒.
1.2.8kFPR1基因反向筛选标记的应用实验 用限制性内切酶PmeI消化pPICZA-EGFP-CYC1TT-kfpr1质粒,将线性化的载体转化至毕赤酵母KO菌株感受态细胞,涂布至YPDZ培养基平板,30 ℃培养2 d.经PCR实验鉴定后,得到重组菌株EGFP-ZeoR-kfpr1-KO.将EGFP-kfpr1-GS115菌株在YPD培养基中连续传代两次后,稀释涂布至YPDR培养基平板上筛选具有雷帕霉素抗性的单菌落,通过测序确认单菌落的基因型(图3).
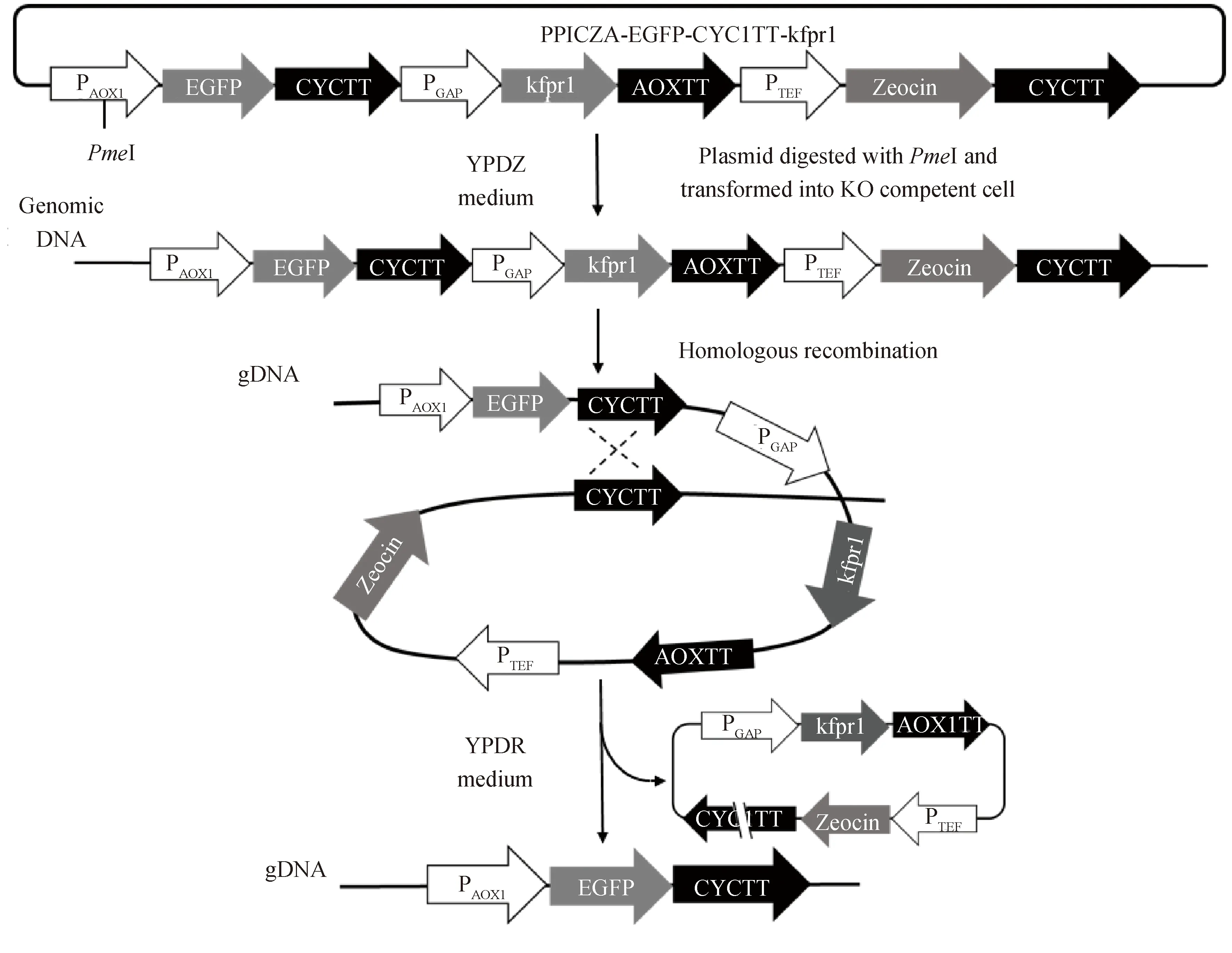
图3 kFPR1基因作为反向筛选标记的应用实验
2 结果
2.1 突变株mut375的分离及表型分析通过转座子突变技术对毕赤酵母GS115菌株进行转座突变,富集得到酵母突变库.经过筛选获得了能够在含有雷帕霉素的培养基生长的突变株mut375.分离单菌落进行培养,在YPDR培养基上生长的情况如图4所示.

图4 mut375在YPDR培养基上的生长情况
2.2 确定mut375突变株转座子插入位点对TAIL-PCR扩增产物测序结果进行分析,利用Internet BLAST在线工具将转座子侧翼序列与毕赤酵母GS115基因组进行比对,发现转座子的插入破坏了PAS_Chr2-2_0375基因(NCBI Gene ID:8199086)开放阅读框的完整性.该基因大小为414 bp,表达产物是由137个氨基酸组成,具有肽基脯氨酰顺反异构酶活性,能够结合FK506免疫抑制类药物.与人(Homosapiens)的FKBP 1B蛋白氨基酸序列具有55.24%的同源性,与酿酒酵母的FPR1蛋白氨基酸序列有76%的同源性,因此将毕赤酵母中的PAS_Chr2-2_0375基因命名为kFPR1.
2.3kFPR1基因缺失菌株KO与功能回补菌株TR的表型结果本研究用毕赤酵母的HIS4基因通过同源重组替换GS115菌株的kFPR1基因(图5A),得到kFPR1基因缺失的突变株KO(表1).通过体外构建pGAPZB-kfpr1表达载体,并整合至菌株KO的染色体上,得到kFPR1基因功能回补菌株TR(表1).kFPR1基因缺失的KO菌株与mut375突变株具有相同表型,均能够在含雷帕霉素的培养基上生长,而功能回补菌株TR则不具备雷帕霉素抗性,与野生型毕赤酵母X33菌株的表型一致(图5B).实验结果表明kFPR1基因的缺失会导致毕赤酵母产生雷帕霉素抗性,回补kFPR1基因的功能会恢复毕赤酵母对雷帕霉素的敏感性.
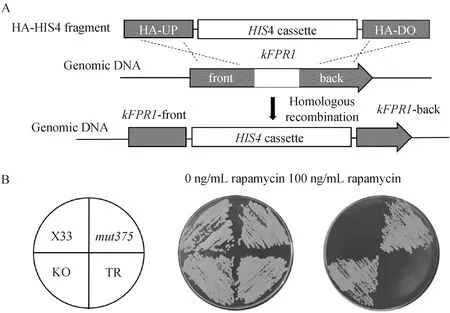
图5 kFPR1基因缺失突变菌株KO与功能回补菌株TRA:毕赤酵母kFPR1基因的敲除.B:突变株mut375、KO及TR菌株在YPDR培养基上的生长情况.
2.4kFPR1基因作为毕赤酵母反向筛选标记的应用将线性化的pPICZA-EGFP-CYC1TT-kfpr1载体整合到毕赤酵母KO菌株的染色体上,使用博来霉素筛选得到重组菌株EGFP-ZeoR-kfpr1-KO.再利用kFPR1基因作为反向筛选标记,使用雷帕霉素筛选得到ZeoR-kfpr1片段丢失的重组菌株EGFP-KO,实现了筛选标记的回收(见图6A).使用甲醇诱导EGFP-KO菌株表达目的蛋白,并在荧光显微镜下观察EGFP的表达情况(见图6B).实验结果显示,本研究以kFPR1作为反向筛选标记,不仅成功将EGFP表达盒成功整合至毕赤酵母基因组中,并且没有引入标记基因,实现了在毕赤酵母中的无标记基因操作.
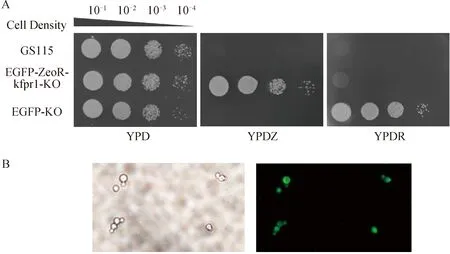
图6 重组菌株EGFP-KO的表型分析A:重组菌株EGFP-ZeoR-kfpr1-KO和EGFP-KO在不同培养基上的生长表型.B:菌株EGFP-KO中绿色荧光蛋白的表达情况.
3 讨论
以毕赤酵母内源性基因kFPR1作为反向筛选标记,博来霉素抗性基因ZeoR作为正向筛选标记,成功将报告基因EGFP整合至酵母染色体,且回收了ZeoR-kfpr1标记单元.实验结果表明kFPR1基因可以作为毕赤酵母的反向筛选标记,为毕赤酵母的无标记基因操作提供了新的工具.
相比于同样适用于毕赤酵母的反向筛选标记mazF,kFPR1的局限性在于目前只适用于毕赤酵母而且需要先对毕赤酵母kFPR1基因进行敲除,其优点在于kFPR1基因产物不影响毕赤酵母的生长代谢.mazF基因产物会抑制毕赤酵母的生长,需要使用诱导型启动子AOX1p严紧调控mazF基因的转录表达,因此反向筛选时需要加入甲醇诱导,以甲醇为唯一碳源时,毕赤酵母的生长速度缓慢.毕赤酵母作为表达外源蛋白的工程菌,常常使用AOX1启动子调控目的基因的表达,会与反向筛选标记mazF的使用产生冲突,影响实验结果.而kFPR1作为反向筛选标记只需要在含雷帕霉素的平板上进行筛选,操作更简便.
本研究开发了一种适用于毕赤酵母的反向筛选标记,实现筛选标记在基因操作过程中的回收以及靶基因的成功整合,克服了筛选标记选择的限制,便于对同一宿主菌株进行多次基因编辑且不会引入抗生素抗性,建立了适用于毕赤酵母的无标记基因操作体系.
