2例先天性胆汁酸合成障碍的临床和遗传学分析
2022-01-03张建霞刘志峰
张建霞 刘志峰
南京医科大学附属南京儿童医院消化科(南京210008)
以胆固醇为原料经一系列酶促反应合成胆汁酸的过程中,任何环节出现障碍均可导致正常胆汁生成减少、中间产物异常堆积,干扰胆汁酸转运过程从而在不同年龄段出现胆汁淤积等不同疾病谱。其中3β-羟基-Δ5-C27-类固醇脱氢酶(3-betahydroxy-delta-5-C27-steroid oxidoreductase)、Δ4-3-氧固醇-5β-还原酶(delta 4-3-oxosteroid 5beta-reductase)参与胆汁酸母核形成的修饰,其缺陷引起的临床表型分别称为先天性胆汁酸合成障碍1 型及2 型,是一类罕见的常染色体隐性遗传疾病,在儿童胆汁淤积性疾病中占比约为1% ~2%[1]。两者临床上常表现为持续性胆汁淤积,于婴儿期甚至新生儿期起病,可伴有脂肪泻或排浅色甚至白陶土样粪便,部分早期进展为肝衰竭。实验室检查中血清及尿液胆汁酸谱均显示为初级胆汁酸水平下降。二者临床表现类似,但在产生的异胆汁酸成分中存在差异,且先天性胆汁酸合成障碍1 型(Inborn errors of bile acid synthesis type 1,IEBAS1)的致病基因为HSD3B7 基因、2 型致病基因为AKR1D1 基因。
因先天性胆汁酸合成障碍较罕见,且无明显特征性临床表型,部分被误诊为胆道闭锁或Citrin 缺陷导致的新生儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by Citrin deficiency,NICCD),然而早期明确诊断、及时药物干预可降低肝脏移植的几率并改善预后,故有必要对其临床特征、基因变异进行总结。
1 临床资料
回顾分析2018年12月至2021年2月南京市儿童医院诊断的IEBAS 患儿的临床资料,共纳入2 例患儿,为无血缘关系家族,分别诊断为IEBAS1、IEBAS2。2 例患儿的家系及基因突变情况见图1、2。
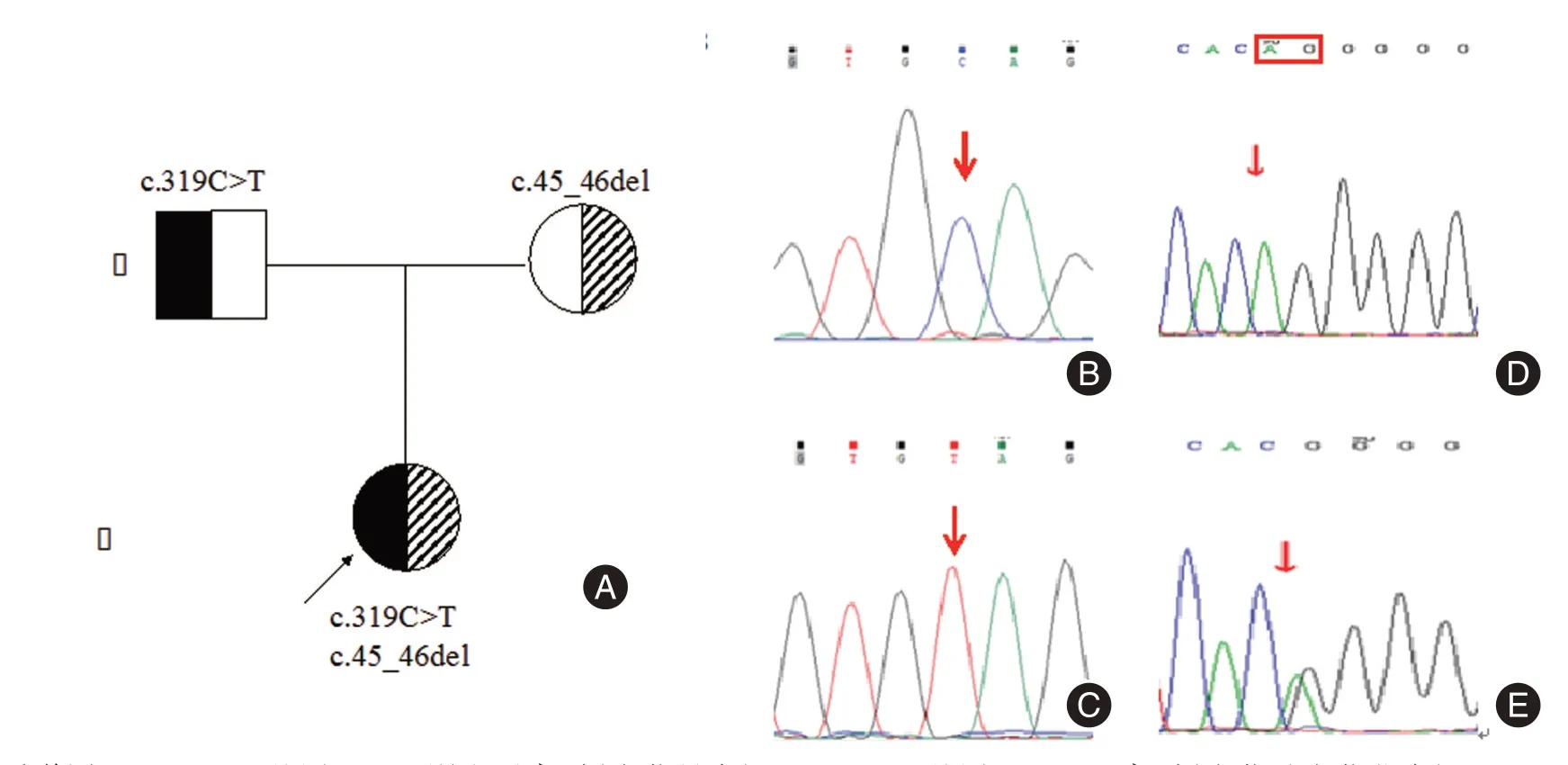
图1 患儿1 的家系及基因突变情况Fig.1 Family pedigree and genetic variants of the patient 1
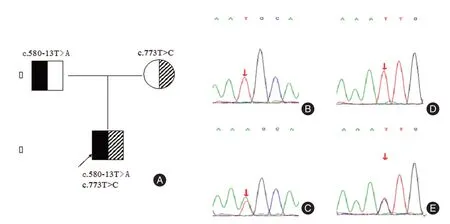
图2 患儿2 的家系及基因突变情况Fig.2 Family pedigree and genetic variants of the patient 2
病例1 患儿4 岁时因“发热3 d”于我院门诊就诊,由于肝脾肿大而转诊于我科门诊,追溯病史患儿新生儿期有黄疸病史,生化检查示胆红素轻度升高,其中以直接胆红素升高为主,凝血酶原时间(prothrombin time,PT)延长,γ-谷氨酰转肽酶降低,影像学显示肝脏饱满,脾脏体积明显增大,脾静脉及门静脉系统明显扩张。
病例2 患儿生后5 d 即出现黄疸,当地医院予口服退黄药物及蓝光治疗后稍有减轻但未完全消退。患儿近4月龄时黄疸进行性加重,于我科住院治疗,血清学检查符合胆汁淤积性肝病:具有黄疸伴肝脏肿大,并有丙氨酸氨基转移酶(alanine aminotransferase,ALT)升高,总胆红素(total bilirubin,TBil)>85 μmol/L 且直接胆红素(direct bilirubin,DBil)/TBil >20%。PT 延长伴γ-谷氨酰转肽酶正常水平,血尿代谢筛查示多种氨基酸及酰基肉碱增高。2 例患儿初诊时血检情况见表1。
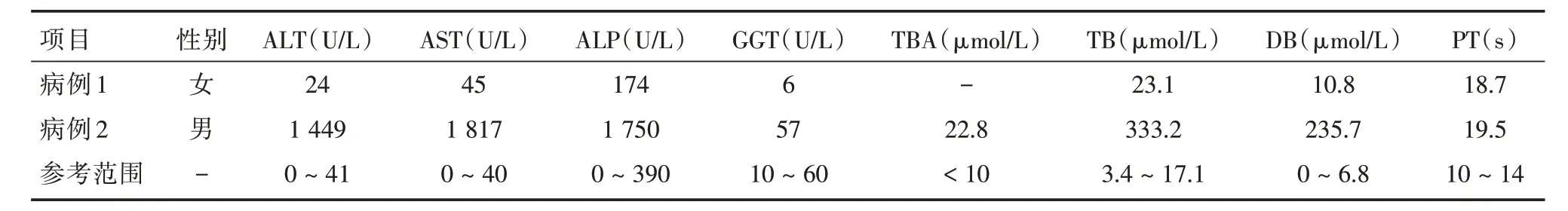
表1 2 例患儿初诊时血检情况Tab.1 Blood test of the two children at initial visit
为明确病因,两患儿均在家长知情同意后进行胆汁淤积症相关基因检测,结果提示病例1 存在HSD3B7 基 因c.45_46del(p.G17Lfs*26)/c.319C >T(p.Q107X)复合杂合突变,病例2 携带AKR1D1 基因c.580-13T >A(splicing)/c.773T >C(p.I258T)复合杂合突变,其中c.319C >T、c.773T >C 均为新发突变。2 例患儿基因检测结果见表2。
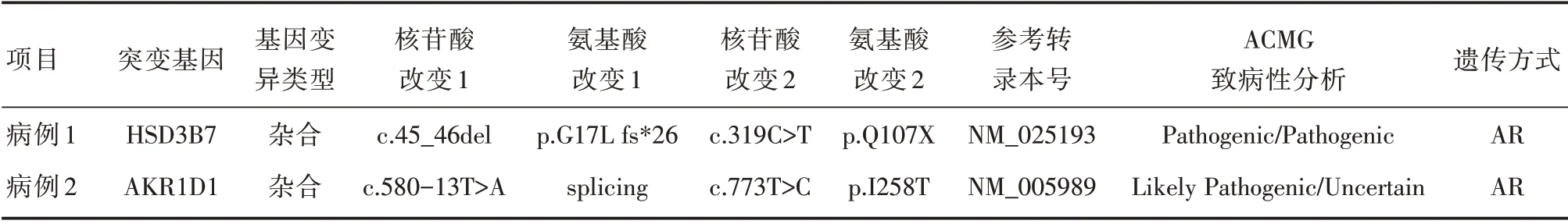
表2 2 例患儿基因检测结果Tab.2 Genetic analysis of the two children
2 讨论
IEBAS1 是先天性胆汁酸合成障碍中最常见的类型,是一种由HSD3B7 基因突变引起的常染色体隐性遗传疾病。该基因定位于染色体16p11.2,编码3β-羟基-Δ5-C27-类固醇脱氢酶,该蛋白主要在肝脏、胰腺和肾脏中表达,另外在心脏、骨骼肌及胎盘中也少量可见[2],主要在胆汁酸合成经典途径中催化7α 羟基胆固醇转化为7α-羟基-4-胆固醇-3-酮,在胆汁酸合成替代途径中催化3β,7α-二羟基-5-胆烷酸转化为7α-羟基3-氧-4-胆烷酸。当该酶缺乏时,积聚的其底物7α 羟基胆固醇可进行侧链修饰,伴或不伴12α 羟基化,从而使得IEBAS1的代谢产物为一系列不饱和胆汁酸,由于不能有效合成初级胆汁酸,致使胆汁循环障碍而导致胆汁淤积[3]。
IEBAS1从新生儿至成人均可发病,新生儿期以黄疸起病,伴有肝脾肿大、脂肪泻等;儿童期可因脂溶性维生素缺乏表现为佝偻病、生长发育迟缓;青少年和成人期以肝硬化起病。实验室检查[4]可见高结合胆红素水平、转氨酶升高、脂溶性维生素缺乏但血清总胆汁酸和γ-谷氨酰转肽酶基本正常。对病人尿液进行快原子轰击质谱(FAB-MS)检测可见3β,7α-二羟基-5-胆烷酸和3β,7α,12α-三羟基-5-胆烷酸等异常胆汁酸。影像学检查可见肝脏肿大伴或不伴脾脏肿大,发展为肝硬化患者可见小结节样肝脏及门静脉高压征,1978年CLAYTON 等[5]首次报道IEBAS1 患者的肝脏病理显示持续性加重的巨细胞肝炎及桥接肝纤维化乃至肝硬化。
本文中提及患儿1 为学龄前儿童,就诊时总胆红素可见轻度升高,PT 稍延长,但转氨酶未见升高,完善影像学检查可见肝脾肿大伴门静脉高压征等肝硬化表现,基因检测提示其存在HSD3B7 基因母源性c.45_46del(p.G17Lfs*26)/父源性c.319C>T(p.Q107X)复合杂合突变,其中移码突变已由CHENG 等[6]报道与3β-羟基-Δ5-C27-类固醇脱氢酶缺陷有关;c.319C >T 为无义突变,目前尚未有文献报道,根据ACMG 指南,该变异初步判定为致病性变异。由于患儿在未采取口服药物治疗前即进展为肝硬化,肝脏移植指征较为明确。
IEBAS2 又称原发性Δ4-3-氧固醇-5β-还原酶缺陷症,其致病基因是AKR1D1 基因,该基因位于染色体7q32-q33,编码Δ4-3-氧固醇-5β-还原酶,此酶为胆汁酸合成经典途径中的关键酶之一。当其发生缺陷时,中间关键产物7α-羟基-及7α,12α-二羟基-4-胆固醇酯-3 酮被催化生成相应的3-氧-5β(H)类似物[7],初级胆汁酸鹅去氧胆酸(chenodeoxycholic acid,CDCA)和胆酸(cholic acid,CA)合成受阻,由于3-氧-5β(H)类似物溶解度低且具有肝毒性,在肝脏异常堆积后造成胆汁淤积。
IEBAS2 常于新生儿期起病,临床表现与IEBAS1 相似但较1 型发展更迅速,早期出现严重胆汁淤积甚至肝衰竭。实验室检查[8-9]可见显著高胆红素血症,以结合胆红素升高为主,ALT、AST、ALP 均轻度升高,脂溶性维生素吸收障碍,γ-谷氨酰转肽酶水平正常或轻度升高,总胆汁酸水平波动于正常范围内。凝血功能检查示凝血酶原时间(PT)和活化部分凝血酶时间(Activated partial thromboplastin time,APTT)明显延长,推测由脂溶性维生素代谢障碍引起。尿液FAB-MS 分析显示以7α-羟基-3-氧代-4-胆烷酸和7α,12α-二羟基-3-氧代-4-胆烷酸为主[10]。B 超检查可出现肝脾不同程度肿大,肝脏穿刺活检示胆管排列紊乱、细胞内胆汁淤积,电镜下可见微绒毛缺失、小胆管裂缝样改变伴致密物沉积[11]。
由于IEBAS 临床表型及生化检查相似,鉴别且明确诊断需结合血尿胆汁酸分析及基因检测。目前通过口服初级未结合胆汁酸如胆酸(CA)、鹅去氧胆酸[12](CDCA)等替代治疗后,IEBAS 临床症状和生化指标可得到明显改善,减少肝脏移植率。研究表明[13],熊去氧胆酸(UDCA)虽可以改善生化、组织学指标,但不能补充人体必需的初级胆汁酸,不能减少因异常胆汁酸而产生的毒性中间代谢产物,因而不推荐使用。对于严重肝功能衰竭或药物治疗无效的患儿,肝脏移植是唯一的办法。
患儿2 IEBAS2 诊断明确,临床表型较为典型,新生儿期即出现进行性黄疸,予茵栀黄口服后黄疸未见明显好转,血清学提示高结合胆红素血症,肝酶明显增高,同时伴有凝血障碍,血尿代谢提示多种氨基酸及酰基肉碱增高,但患儿胆汁酸检测高于参考范围,与既往文献报道不符,考虑与检测前已口服熊去氧胆酸治疗,增加了胆汁酸分泌相关。未明确诊断前患儿口服UDCA 治疗,初期可见黄疸轻度好转,肝酶明显下降,但后期出现黄疸反复、转氨酶复升,完善基因检测提示患儿携带AKR1D1 基因c.580-13T >A(splicing)/c.773T >C(p.I258T)复合杂合突变,其中剪接突变CHEN 等[14]已报道与Δ4-3-氧固醇-5β-还原酶缺陷有关,而源自母亲的c.773T >C 尚未被已有文献提及,其在人群数据库中为低频变异,根据ACMG 指南,该变异初步判定为临床意义不明,其功能仍需进一步验证。
本文总结了两例先天性胆汁酸合成障碍的临床表型及基因型,扩展了HSD3B7、AKR1D1 基因突变谱,其中两新发突变的具体致病机制仍有待进一步研究。先天性胆汁酸合成障碍是一种罕见的遗传性胆汁淤积疾病,血尿胆汁酸成分分析及基因检测可进行鉴别及确诊,口服UDCA 可短期改善患儿症状,但长期使用效果欠佳,推荐明确诊断后,尽早口服初级胆汁酸以改善患儿生存质量及预后,若药物治疗病情仍进一步加重,肝脏移植为最后一道防线。
