Cr、Mn、Co、Ni掺杂对Al13Fe4相稳定性和力学性能影响的第一性原理研究
2021-12-27史志铭王存权吴玉婷张江超
庞 娜, 史志铭, 王存权, 吴玉婷, 张江超, 孙 江
(内蒙古工业大学 材料科学与工程学院, 呼和浩特 010051)
1 引 言
铝铁基铝合金具有热稳定性好、热膨胀小、弹性模量高、耐腐蚀性好等优点,在各种机械和高温环境中具有良好的应用前景. 这些性能归因于α-Al基体中细小、坚硬、耐热的Al13Fe4相,增加合金中Fe含量可进一步改善这些性能[1]. 但随着Fe含量的增加,容易形成粗大的片状、针状和板条状的Al13Fe4相,严重制约了Al-Fe合金的力学性能和抗蠕变能力[2]. 因此,需要找到一种细化和改善Al13Fe4相形貌和分布的有效方法.
固溶强化作为提高Al13Fe4相韧性、改善形貌和分布的重要途径,广泛应用于Al-Fe合金中. Ti、Cr、Mn、Co、Ni、Cu、Zn等过渡族元素由于在铝合金中的固溶性和扩散速率较低而形成非常稳定且抗粗化的金属间化合物,常作为掺杂元素加入,可以有效地提高Al-Fe合金的热稳定性、机械强度、高温机械性能[3]. 研究发现,三元Al-Fe-X合金(X: Zr, Mo, Si, Ni, Cr)在室温和400 ℃高温时均表现出较高的强度. 加入Ni可以细化Al13Fe4相,促进Al3Ni和Al9FeNi相的形成,提高Al-Fe合金的高温力学性能和弹性模量,降低其热膨胀系数[4]. Mn可以稳定亚稳的Al6Fe相,形成Al6(Fe, Mn)连续固溶体,提高了合金的屈服强度和抗拉强度[5]. 添加0.2wt% Co完全固溶于Al13Fe4相,可以明显改善Al-5Fe合金中Al13Fe4相的形貌[6]. 过渡族金属在α-Al基体中的固溶度很低,可以溶解在Al13Fe4相中形成置换固溶体. 例如,Ni、Mn、Cr、Co元素在Al13Fe4相中的固溶度分别达到3-4at%(也有报道高达7at%[7])、4-5%at、4at%和12.4at%[8].
目前在运用理论方法和第一性原理方法方面已研究了纯Al13Fe4的结构、电子和磁性、催化表面以及其内在缺陷[9-11]. 然而,对于过渡族元素固溶于Al13Fe4相的研究却很少. 尚未清楚不同掺杂浓度的合金元素对Al13Fe4相的影响规律. 特别是合金元素细化Al13Fe4相的理论研究仍然缺乏. 此外,Al13Fe4相的力学性能对Al-Fe合金的力学性能有很大的影响. 过渡族元素的固溶会引起Al13Fe4相的稳定性和力学性能的变化. 然而,这些结果在实验上很难得到,因此需要在理论计算的基础上从微观电子结构和弹性理论出发,研究相稳定性、脆塑性等性能. 本文基于第一性原理系统的研究了Cr、Mn、Co、Ni加入对Al13Fe4相结构、电子和机械性能的影响,分析了过渡族元素对Al-Fe合金的强化机理.
2 晶胞模型及计算方法
Al13Fe4是Al-Fe合金的主要相成分,属于单斜晶系,空间群为C2/m[12]. Al13Fe4单胞包含15种Al和5种Fe的晶体学位置,共有102个原子(78个Al和24个Fe原子),单胞结构如图1所示. Cr、Mn、Co、Ni与Fe具有相似化学性质,更有利于占据Al13Fe4相的Fe位[7,9,13,14]. 在Al13Fe4单胞中分别加入1、2和4个Cr、Mn、Co、Ni原子置换5个不同晶体学位置的Fe原子,其中在加入2和4个原子时只考虑置换对称位置的Fe,置换后形成掺杂浓度分别为0.98 at%、1.96at%和3.92at%的Al78(Fe24-xMx) (M = Cr、Mn、Co、Ni;x = 1、2、4).
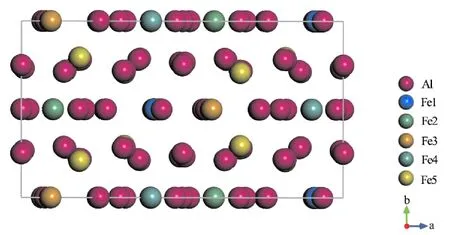
图1 Al13Fe4单胞沿[010]方向的投影Fig. 1 Projection of the Al13Fe4 unit cell along the [010] direction
本文基于密度泛函理论(DFT),采用CASTEP量子力学模块进行计算. 采用广义梯度近似方法(GGA),电子关联选取PBE泛函. 基于收敛性测试,Al13Fe4和Al78(Fe24-xMx)化合物的布里渊区k点均设为2×3×2,平面波截断能取400 eV. 采用自洽场(SCF)法计算总能量,自洽场计算基于Pulay密度混合法. 收敛标准为电子弛豫最大能量变化低于10-5V/atom,作用于原子的最大应力小于0.03 eV/Å. 由于Al13Fe4属于复杂金属间化合物不考虑自旋极化[9].
Al78(Fe24-xMx) 合金的形成焓定义如下:
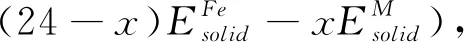
(1)

3 结果与讨论
3.1 Al13Fe4相稳定性分析
添加过渡族元素通过形成置换固溶体而影响Al13Fe4相的形成焓. 首先,对Al13Fe4晶体进行结构优化,以保证计算的可靠性. 优化后Al13Fe4晶体的晶格参数为a= 15.366 Å,b= 8.012 Å,c= 12.393 Å,β= 107.67°,与实验值(a= 15.487 Å,b= 8.0831 Å,c= 12.476 Å,和β= 107.72°)的误差很小,证明计算参数设置合理[12].
图2为不同掺杂浓度的Cr、Mn、Co、Ni置换不同Fe位后Al78(Fe24-xMx)的形成焓,灰色虚线为Al13Fe4的形成焓. Al78(Fe24-xMx)的形成焓均为负,表明这些化合物能够形成,其晶体结构是热力学稳定的. 形成焓越低,对应的晶体结构越稳定. 可以发现,与Al13Fe4的形成焓相比,加入Ni和Co原子提高了化合物的稳定性,而加入Cr和Mn原子降低了化合物的稳定性. 随着掺杂浓度的增加,Al78(Fe24-xNix)和Al78(Fe24-xCox)的稳定性增加,而Al78(Fe24-xCrx)和Al78(Fe24-xMnx)的稳定性降低. 相同掺杂浓度下化合物的形成焓依次为:Al78(Fe24-xCrx) > Al78(Fe24-xMnx) > Al13Fe4> Al78(Fe24-xNix) > Al78(Fe24-xCox).
Cr和Mn更倾向于占据Fe-5位置,而Co和Ni更倾向于占据Fe-1位置. 与Fe相比,Co和Ni的3d电子轨道含有较少的电子,有利于占据配位数较大的Fe-1位,而Cr和Mn则占据配位数较小的Fe-5位. 可以推断,过渡族元素3d电子轨道中的电子数对择优占位有重要影响. 有研究表明不同位置的Fe在费米能级上的贡献是不同的,这与配位数的不同有关,原子间键合过程是放热的,因此配位数越大,键合度越高,稳定性越好[15]. Co和Ni的加入降低了形成焓,占据了配位数较大的Fe-1位置.
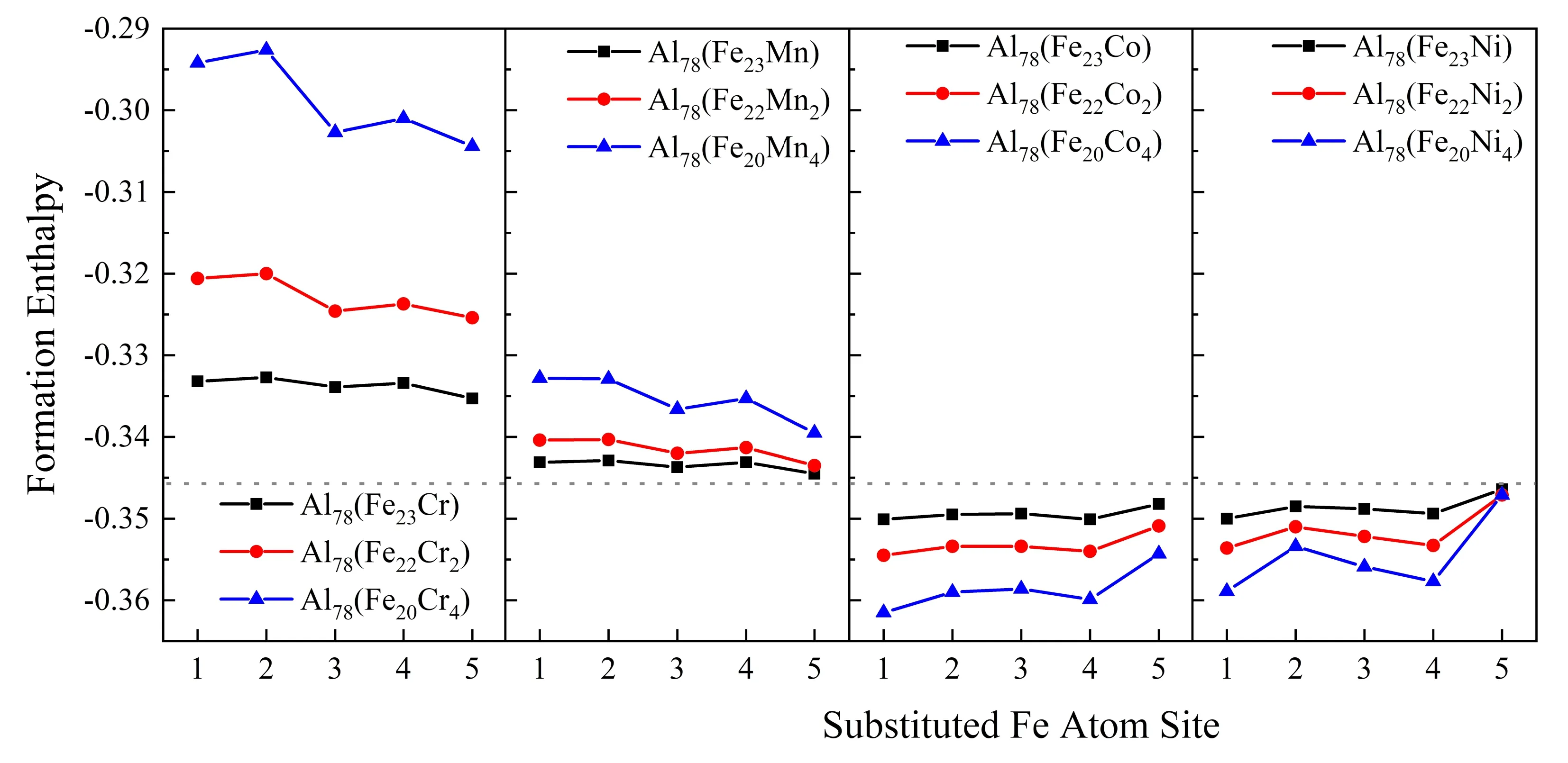
图2 不同置换位置和掺杂浓度Al78(Fe24-xMx)化合物的形成焓Fig. 2 The values of formation enthalpy of Al78(Fe24-xMx) compounds with different substituted sites and different doping concentrations
Al13Fe4和Al78(Fe24-xMx)的单胞体积如图3所示. 灰色虚线表示Al13Fe4的单胞体积. 可以看出,过渡族元素掺杂引起了晶格畸变,随着过渡族元素的增加,Al78(Fe24-xMx)的体积增加. 化合物体积的变化趋势与M原子半径的变化趋势基本一致. 在相同掺杂浓度下,同一元素在稳定性更高的置换位置的体积最小,结构的畸变也最小. 值得注意的是,仅考虑原子半径对体积的影响,Al78(Fe24-xCox)的体积应大于Al78(Fe24-xNix); 然而,这与目前的结果相反. 因此,与原子半径相比,形成焓对晶胞体积的作用更为突出. 这是因为Al13Co4和Al13Fe4都属于单斜晶系,具有相同的晶体结构,可以形成连续的Al13(Fe, Co)4固溶体[7],与Al13Fe4相比,Al13Co4的形成焓更低[9]. 这一特征在其他过渡族元素中没有观察到. 过渡族元素的加入虽然不同程度地改变了Al13Fe4的晶格常数,但是这种变化很小,不足以引起晶体结构对称性的变化,从而为使用原始晶格体系计算力学性能提供了理论依据. 在确定优先占位的基础上,研究了Cr、Mn、Co、Ni对Al13Fe4相其它性能的影响.
在护理专业发展过程中,教师发挥着重要的作用,是护理专业学生前进路上的引导者。但是就目前护理专业的现状来看,高职院校护理专业教学中护理教师因为一直处于校园这一封闭化的氛围中,其学习和传授的护理知识也是偏向于学术化,在一定程度上与当前的教育信息化时代相脱离。相比于一线护理人员,其对医院护理岗位信息化以及信息化操作人才的需求了解较少。这一教学现状严重影响了护理专业教学中信息化教育的渗透,阻碍了护理专业教育信息化的发展,不利于实现护理专业毕业生职业能力的提升。
3.2 力学性能分析
Al13Fe4具有13个独立弹性常数. 利用文献[16]中给出单斜体系在0 GPa时的力学稳定性判据判断Al13Fe4结构的稳定性. 剪切模量G代表塑性变形抗力,体积模量B代表断裂抗力. 根据得到的弹性常数,B和G可以通过Voigt-Reuss-Hill (VRH)方法[17]估计.
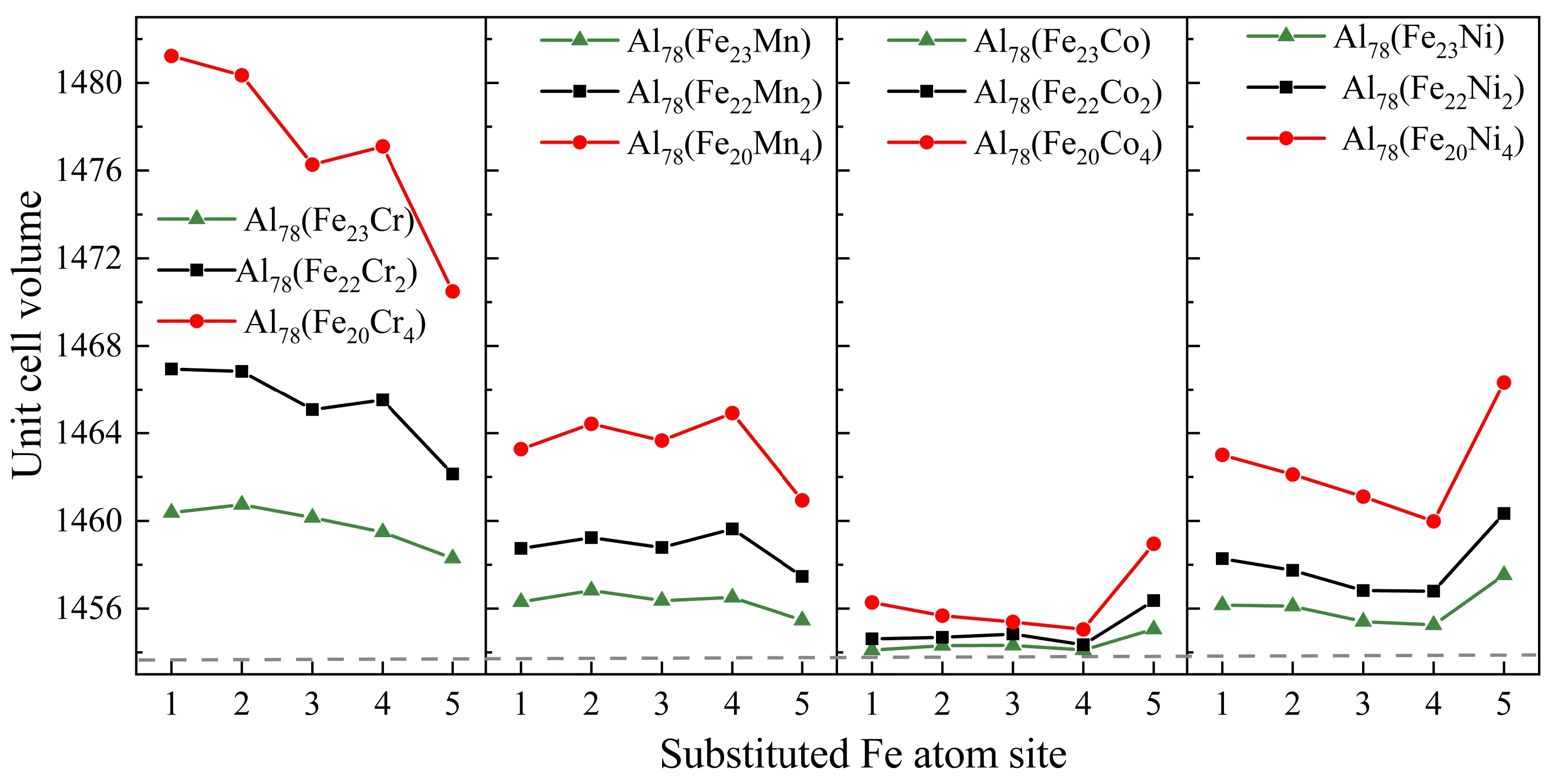
图3 Al78(Fe24-xMx)化合物的单胞体积变化Fig. 3 The unit cellvolumes of Al78(Fe24-xMx) compounds
表1为Al13Fe4和Al78(Fe24-xMx)化合物的弹性常数计算结果. 值得注意的是,M部分置换Fe破坏了晶体的局部对称性,相应地使晶体的晶格对称性由单斜变为三斜. 然而,计算表明掺杂的晶体与纯相的偏差很小,因此掺杂体系可以被视为单斜晶系,所得弹性常数均满足力学稳定判据. 计算得到的Al13Fe4的弹性常数与文献[18]的DFT计算基本一致.
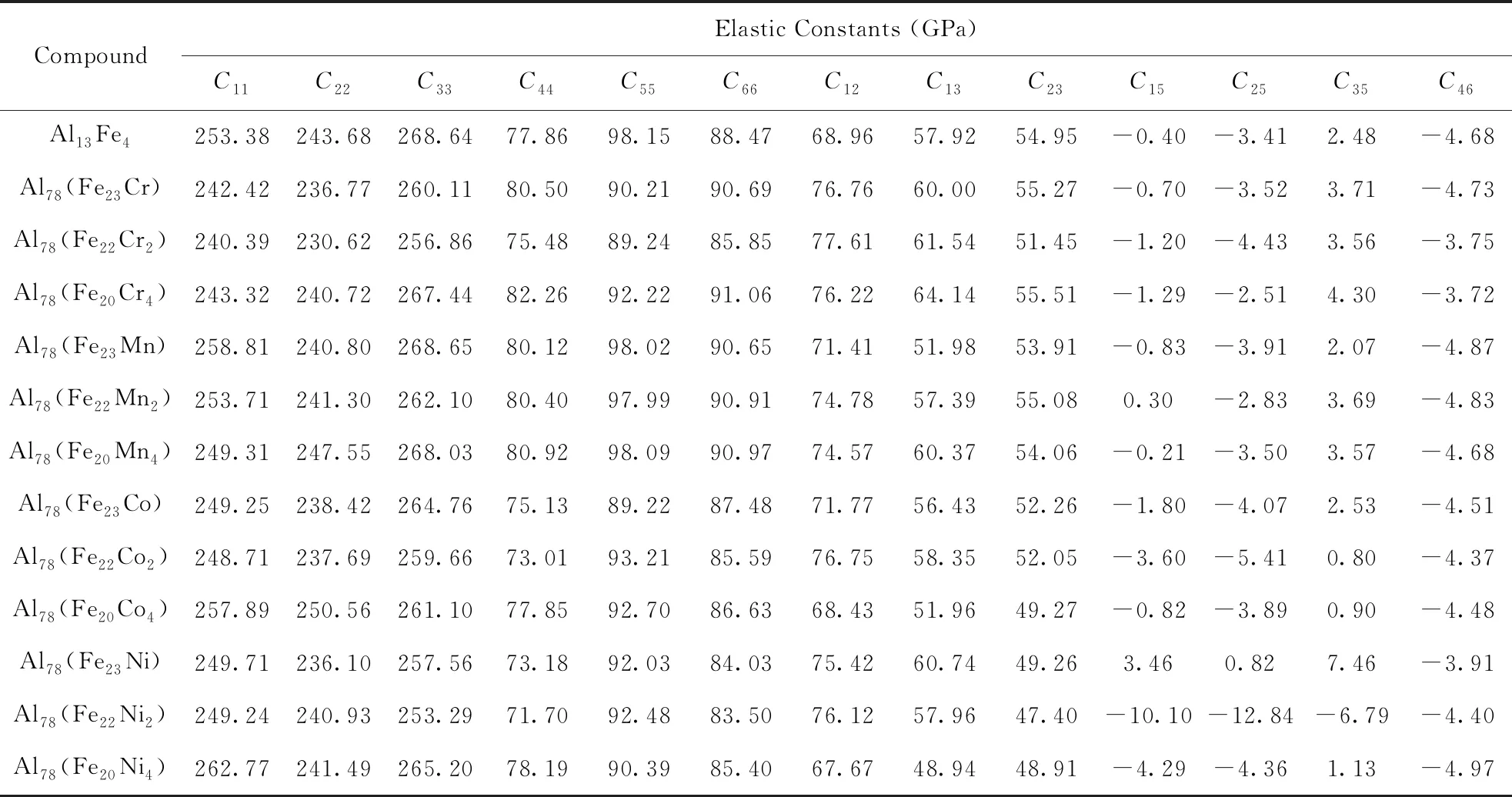
表1 Al13Fe4和Al78(Fe24-xMx)的弹性常数
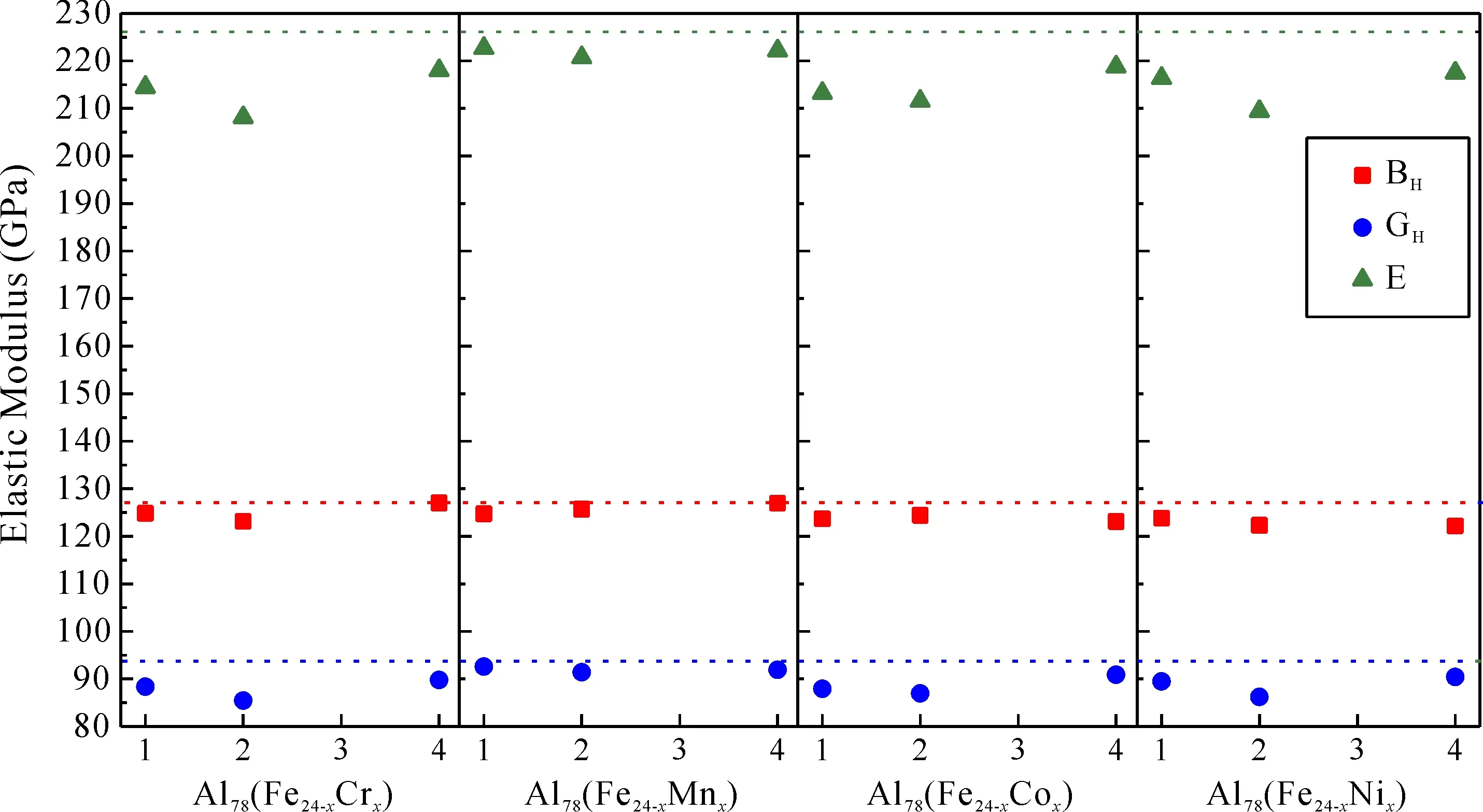
图4 Al13Fe4和Al78(Fe24-xMx)的弹性模量Fig. 4 Elasticmoduli of Al13Fe4 andAl78(Fe24-xMx)
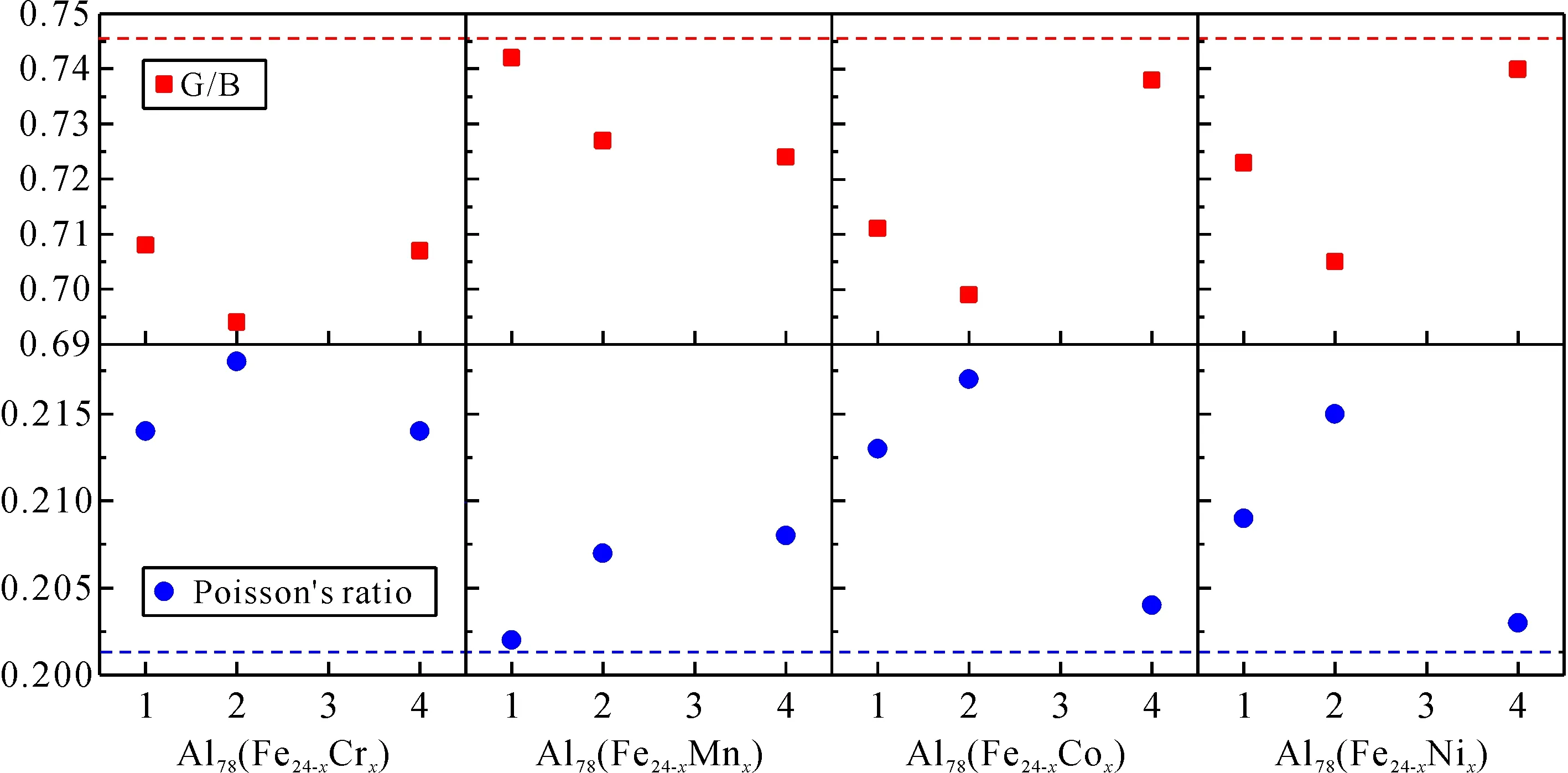
图5 Al13Fe4和Al78(Fe24-xMx)的G/B值和泊松比 Fig. 5 G/B values and Poisson’s ratios of Al13Fe4 and Al78(Fe24-xMx)
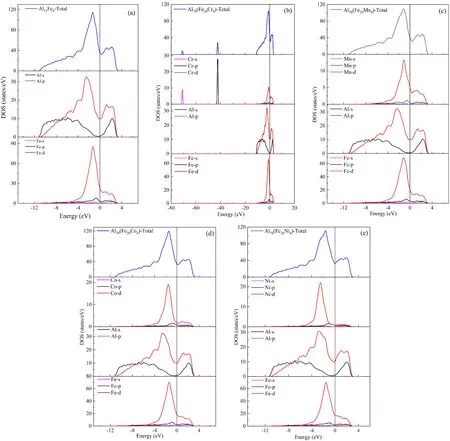
图6 Al13Fe4和Al78(Fe20M4)的态密度(DOSs)Fig. 6 Densities of states (DOSs) of Al13Fe4 and Al78(Fe20M4)
弹性模量的计算值见图4. 虚线表示Al13Fe4相对应的值. 与Al13Fe4相比,Al78(Fe24-xMx)的G、B、E值均有不同程度的降低,塑性变形能力有所增强. 同一元素不同掺杂浓度下,置换2个Fe原子即掺杂浓度为1.96at%时对弹性性能的影响最大. 与Al13Fe4相比,Al78(Fe24-xMx)相和α-Al基体的弹性匹配度更高. 室温原位拉伸试验表明,裂纹主要在Al13Fe4粗颗粒内部形成[19]. M原子的加入有助于防止裂纹的扩展.
图5为Al13Fe4和Al78(Fe24-xMx)的G/B值和泊松比,x轴表示每个晶胞所取代的原子数.根据Pugh的准则,剪切模量与体积模量的比值提供了一种衡量延性-脆性行为的方法[20].G/B值越高,脆性越大;G/B值越低,塑性越好,其临界值为0.57. 此外,当泊松比大于0.26时,材料具有延性;否则为脆性. Al13Fe4和Al78(Fe24-xMx)化合物的G/B值均大于0.57,故它们都表现出一定的脆性. 但是合金化可以提高这些金属间化合物的延性. Cr对改善化合物力学性能作用更明显,而Mn的影响较小. 这些金属间化合物的泊松比都在0.1 ~ 1/3之间,表明它们是脆性的,同时存在金属键和共价键. 随着掺杂浓度的增加,添加M对脆性的改善先增大后减小.
3.3 电子性质分析
为了进一步了解Cr、Mn、Co、Ni在Al13Fe4相中的作用,研究了Al13Fe4和Al78(Fe20M4)的电子结构,如图6和图7所示. 由于掺杂浓度不会改变态密度(DOS)的趋势,为了简化分析,只对Al78(Fe20M4)化合物进行了分析. 由于M原子置换比例小,且这些元素的性质相似,Al13Fe4和Al78(Fe20M4)的DOS非常相似. Al13Fe4和Al78(Fe20M4)电子结构的主要特征是杂化,费米能级附近的成键峰主要来源于Fe-3d态和Al-3p态的贡献,共价键与金属键共存,存在赝能隙,有利于提高金属间化合物的结构稳定性. 费米能级(EF)位于赝能隙底部靠左的位置,表明Al13Fe4是一种高熔点的金属间化合物,且化合物的键合状态没有完全被占据,化合物具有一定的金属性.
此外,由于共价键的键合强度和方向性较大,化合物整体表现为脆性. 费米能级的N(EF)能值与其结构稳定性之间存在一定的相关性,即N(EF)值越低,稳定性越高. 从图7可以看出,这些N(EF)值依次减小:Al78(Fe20Cr4) > Al78(Fe20Mn4) > Al13Fe4> Al78(Fe20Ni4) > Al78(Fe20Co4). 这与形成焓的研究结果一致.
3.4 强化机理的讨论
已有实验研究表明,Cr、Mn、Co、Ni可以细化Al-Fe合金的晶粒,提高合金的性能. 通过对Al13Fe4相的相稳定性、力学性能和电子结构的研究,可以预测Al-Fe合金中过渡族元素的强化机理.
根据相变的热力学驱动力,等压条件下的吉布斯自由能可表示为G(t)=H(t)-TS(t).Al13Fe4相的结晶需要满足ΔG<0 (ΔG=ΔH-TΔS).在相同条件下当M原子置换Fe原子时,Al78(Fe24-xMx)的ΔS可视为变化相等,Co和Ni原子取代后化合物的形成焓降低,这意味着增加了结晶驱动力,更有利于促进Al13Fe4相的形核和细化. 在Cr、Mn、Co、Ni元素中,Ni对改善Al13Fe4相形貌和细化晶粒的效果最好. 此外,稳定性的降低可以促进Al-Fe合金中其它亚稳相的保留,如Al6(Fe,M). 实验中在Al-Fe合金中加入Cr和Mn原子后观察到亚稳AlxFe相的存在[9].
4 结 论
采用第一性原理计算方法研究了过渡族元素(Cr、Mn、Co和Ni)对Al13Fe4金属间化合物结构稳定性、力学性能和电子结构的影响. 主要研究结果如下:

图7 Al13Fe4和Al78(Fe20M4)的总态密度(TDOSs)Fig. 7 Total densities of states (DOSs) of Al13Fe4 and Al78(Fe20M4)
(1)相同掺杂浓度化合物的形成焓按如下顺序减小:Al78(Fe24-xCrx) > Al78(Fe24-xMnx) > Al13Fe4> Al78(Fe24-xNix) > Al78(Fe24-xCox). Cr和Mn更倾向于占据Fe-5位,而Co和Ni更倾向于占据Fe-1位. 总的来说,形成焓的降低增加了Al78(Fe24-xNix)和Al78(Fe24-xCox)相的相变驱动力,更有利于促进Al13Fe4相的形核和细化. 此外,Al78(Fe24-xCrx)和Al78(Fe24-xMnx)的稳定性降低,促进了Al-Fe合金中Cr和Mn原子取代后形成其他亚稳相,如Al6(Fe,M).
(2)Cr、Mn、Co和Ni可以改善金属间化合物的脆性,增强塑性变形能力. 随着掺杂浓度的增加对脆性的改善呈现先增大后减小的趋势. Al13Fe4金属间化合物的脆性是由于晶体内Fe-3d态和Al-3p态的强共价相互作用造成的. 合金化削弱了Fe-3d电子和Al-3p电子的共价相互作用,从而提高了塑性.
(3)计算为进一步了解Al-Fe合金的合金化强化机理提供了理论依据.
