川芎药材与饮片指纹图谱对比研究
2021-12-27瞿昊宇谢梦洲陈光宇
瞿昊宇,何 群,谢梦洲,3,4,5,陈光宇,3,4,5*
(1.湖南中医药大学 信息科学与工程学院;2.湖南中医药大学 湖南省药食同源功能性食品工程技术研究中心;3.湖南中医药大学 中医诊断学湖南省重点实验室;4.湖南中医药大学中医心肺病证辨证与药膳食疗重点研究室;5.湖南中医药大学 中医学院,湖南 长沙410208)
川芎为伞形科植物川芎Ligusticum chuanxiongHort.的干燥根茎,主产于四川省彭州、都江堰等地,为四川道地药材(道地药材是指具有特定产区、货真质优的中药材)。川芎功能与主治为活血行气、祛风止痛,用于胸痹心痛、胸胁刺痛、跌扑肿痛、月经不调、经闭痛经、癥瘕腹痛、头痛、风湿痹痛[1],为历版《中华人民共和国药典》收载(如2015版《中华人民共和国药典》一部40-41页),经炮制后以饮片入药。现代化学和药理研究表明,川芎的活性成分主要包括挥发油、酚酸类、生物碱类和内酯类等,其中,阿魏酸是川芎中重要的活性成分,是评价川芎质量的重要标准之一[2]。现已发现阿魏酸具有抗动脉粥样硬化,抗血小板凝集和血栓,清除亚硝酸盐、氧自由基、过氧化亚硝基,以及抗菌消炎、抗肿瘤、抗突变、增强免疫功能等作用[3]。早期曾从国产川芎分离出有特殊气味、易挥发的油状生物碱、阿魏酸、酚性物质和挥发油等,近年来,对川芎化学成分的研究比较深入,已分离鉴定出了260余种成分[4]。作为临床常用中药,有关川芎化学成分的研究较多,国内外均有大量研究报道[5]。一般认为,川芎经炮制后,质变酥脆,利于粉碎和有效成分煎出。2015版《中华人民共和国药典》一部“川芎药材及饮片”项下规定了性状、鉴别、检查、浸出物、含量测定,未规定指纹图谱检测。但仅从已有的检测项目(如浸出物、阿魏酸含量等)无法全面控制川芎药材质量,研究其单体成分并不能洞悉川芎功效的全貌,不能全面反映川芎炮制前后质量属性的改变,从而影响川芎药材及饮片质量控制及质量评价。目前,国内外已有相关文献报道川芎药材或饮片的指纹图谱研究,但其方法未被药典收载。且其指纹图谱未对原料来源进行聚类分析,对于量质传递研究还缺乏依据。本研究基于液相色谱法,对文献报道的川芎指纹图谱研究进行验证,为药典收载方法提供重现性依据。根据指纹图谱验证实验结果,对川芎指纹图谱峰进行辨认,为川芎药材-饮片量值传递提供实验依据。
1 仪器与药材
1.1 仪器
Ultimate 3000高效液相色谱系统(赛默飞世尔科技(中国)有限公司;包括G1316A四元梯度泵,G1313A标准型自动进样器,G1316A恒温箱,G1314A紫外检测器,Agilent Chemstation色谱工作站);中文液晶台式超声波清洗器(昆山美美超声仪器有限公司);ME204E型电子分析天平(上海梅特勒-托利多仪器有限公司);TCS-100型电子台秤(上海乾峰电子仪器有限公司);PW135型中草药粉碎机(天津泰斯特仪器有限公司);SHH.W21电子三用水箱(北京中兴伟业仪器有限公司);QY-1中药切片机(温州顶历医疗器械有限公司);101-0AB型电热鼓风干燥箱(北京中兴伟业仪器有限公司)。
1.2 药材及试剂
各产地、批号的川芎药材经湖南中医药大学王智老师鉴定为川芎Ligusticum chuanxiongHort.的干燥根茎;阿魏酸对照品(批号:110773-201614),购自中国食品药品检定研究院(国家药品标准物质),供含量测定用(不少于99.0%50 mg);甲醇、乙腈为色谱纯,水为重蒸馏水;液相色谱柱(月旭公司UltimateⒸXB-C18型,5μm,4.6mm×250mm)。
2 实验方法与结果
2.1 川芎药材指纹图谱测定方法
2.1.1 色谱条件与系统适用性试验[1-6]液相色谱柱以十八烷基硅烷键合硅胶为填充剂,以甲醇(A)-0.1%磷酸水溶液(B)为流动相;梯度洗脱(0~10 min,15%→30%A;10~20 min,30%→50%A;20~40 min,50%→70%A;40~45min,70%A;45~55 min,70%→100%A;50~60 min,100%A;60~65min,100%→15%A;65~70 min,15%A)流速:0.8 m L·min-1,检测波长为270nm,柱温:30℃,进样量:10μL,理论板数按阿魏酸峰计算应不低于2 000。
2.1.2 阿魏酸对照品溶液制备 取阿魏酸约10mg,精密称定,置于10 m L棕色容量瓶中,精密加入70%甲醇定容至刻度,摇匀,制成每1m L含阿魏酸1.13mg的阿魏酸对照品浓溶液1,精密移取1 m L阿魏酸对照品浓溶液1置10 m L棕色容量瓶中,加70%甲醇定容至刻度,摇匀,制成每1 m L含阿魏酸0.113mg的对照品浓溶液2,精密移取2 m L阿魏酸对照品浓溶液2于10 m L棕色容量瓶中,加70%甲醇定容至刻度,摇匀,制成每1 m L含阿魏酸22.6μg的对照品溶液,过0.22μm微孔滤膜至进样小瓶中,即得。
2.1.3 供试品溶液制备基本方法 取川芎粉末(过四号筛)约0.5 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50m L,密塞,称定重量,置超声波提取器中超声60 min(200 W,50 Hz),放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,静置,取10 m L上清液离心7 min,取上清液,过0.22μm的微孔滤膜至进样小瓶中,即得供试品溶液。
2.1.4 阴性对照供试品溶液的制备 取缺川芎药材的样品,按“2.1.3”法同法制备,即得阴性供试品溶液。
2.2 方法学考察[7-10]
2.2.1 精密度试验 取同一供试品溶液,连续进样6次,检测指纹图谱峰,各共有峰相对保留时间与峰面积比值大体一致,阿魏酸色谱峰面积RSD=1.382%(n=6),表明仪器精密度良好。
2.2.2 重复性试验 取同一批样品6份,按供试品溶液的制备法制备成供试品溶液,按“2.1.1”项下色谱条件分别进样,测定其色谱图,各共有峰相对保留时间与峰面积比值大体一致,阿魏酸色谱峰面积RSD=2.272%(n=6),表明本分析方法重复性良好。
2.2.3 稳定性试验 取同一供试品溶液,分别于0、4、8、12、24、48 h进样,测定其HPLC谱图,各共有峰相对保留时间和峰面积比值基本一致,阿魏酸色谱峰面积RSD=2.772%(n=6),表明本分析方法稳定性良好,供试液在48h内稳定。
2.3 川芎药材及饮片指纹图谱建立
根据CX-02、06、07、08、09、17、20、21、22、23、24、25、26、29、30等15批次川芎药材及饮片色谱图,参照文献报道方法进行匹配,15批药材指纹图谱见图1,15批饮片指纹图谱见图2,生成的川芎药材对照特征图谱见图3,川芎饮片对照特征图谱见图4。阴性对照品溶液(甲醇空白)见图5,阿魏酸对照品溶液见图6,川芎药材供试液指纹图谱见图7,川芎饮片供试液指纹图谱见图8,各批药材供试品对照特征图谱相似度见表1。各批饮片供试品对照特征图谱相似度见表2。
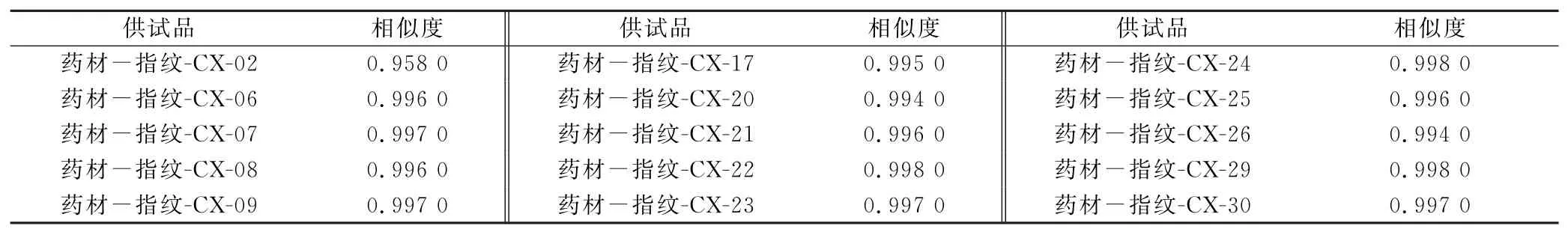
表1 15批川芎药材HPLC指纹图谱相似度

表2 15批川芎饮片HPLC指纹图谱相似度
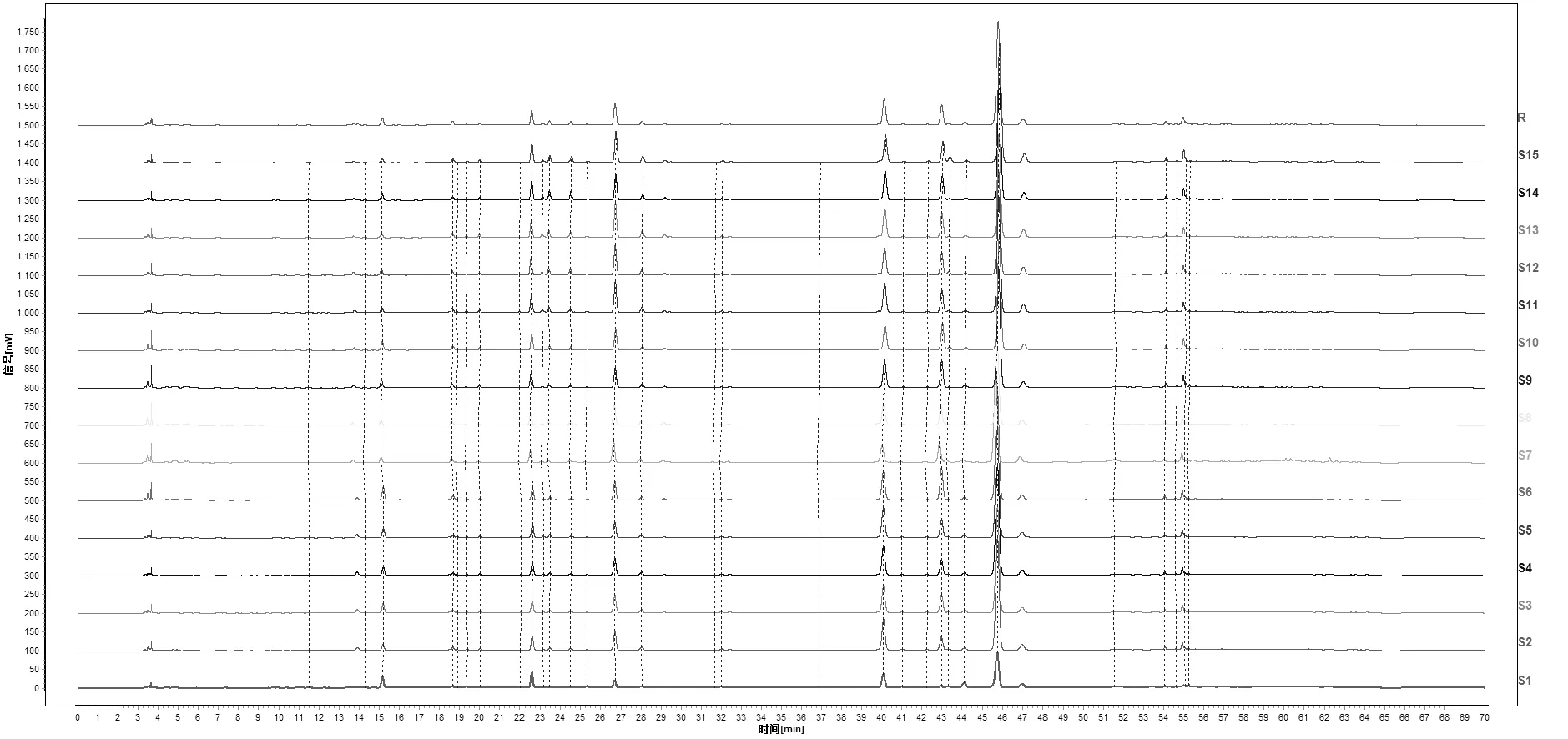
图1 15批川芎药材指纹图谱

图2 15批川芎饮片指纹图谱

图3 川芎药材对照特征图谱
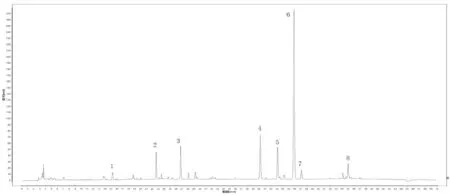
图4 川芎饮片对照特征图谱
图5 阴性对照品指纹图谱(甲醇溶剂空白)

图6 阿魏酸对照品指纹图谱
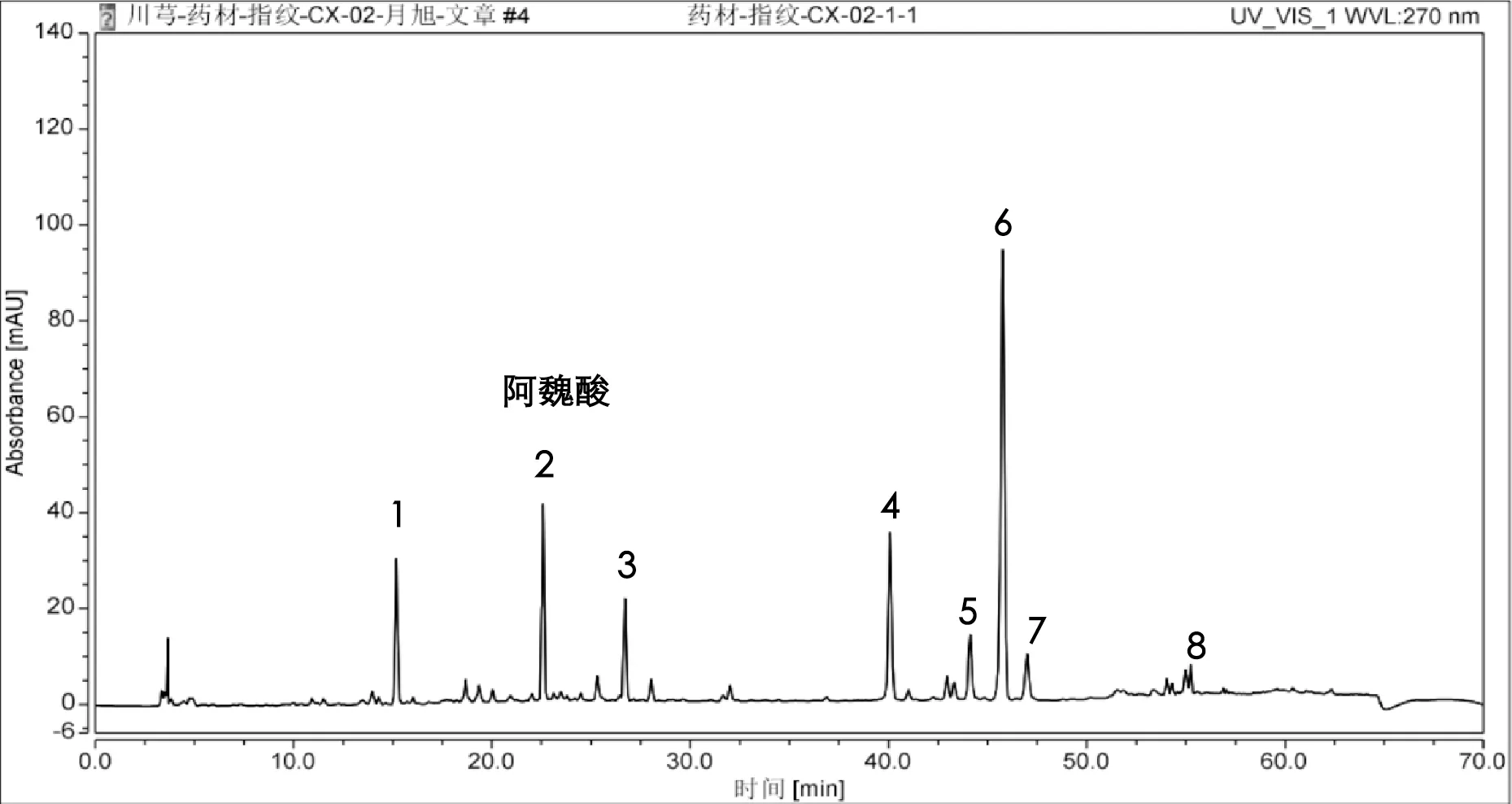
图7 川芎药材指纹图谱

图8 川芎饮片指纹图谱
由表1知,川芎药材各指纹图谱相似度值皆不低于0.9, 故质量标准应规定川芎药材指纹图谱相似度不低于0.9。
由表2知,川芎饮片各指纹图谱相似度值皆不低于0.9,故质量标准应规定川芎饮片指纹图谱相似度不低于0.9。
2.4 聚类分析
以川芎药材(30个共有峰)及饮片(29个共有峰)15批样品中所有共有峰面积为变量,利用DPS 7.05软件中“系统聚类”方法对川芎药材及饮片HPLC指纹图谱进行聚类分析,选择数据不转换、欧氏距离、最短距离,结果见图9、图10。
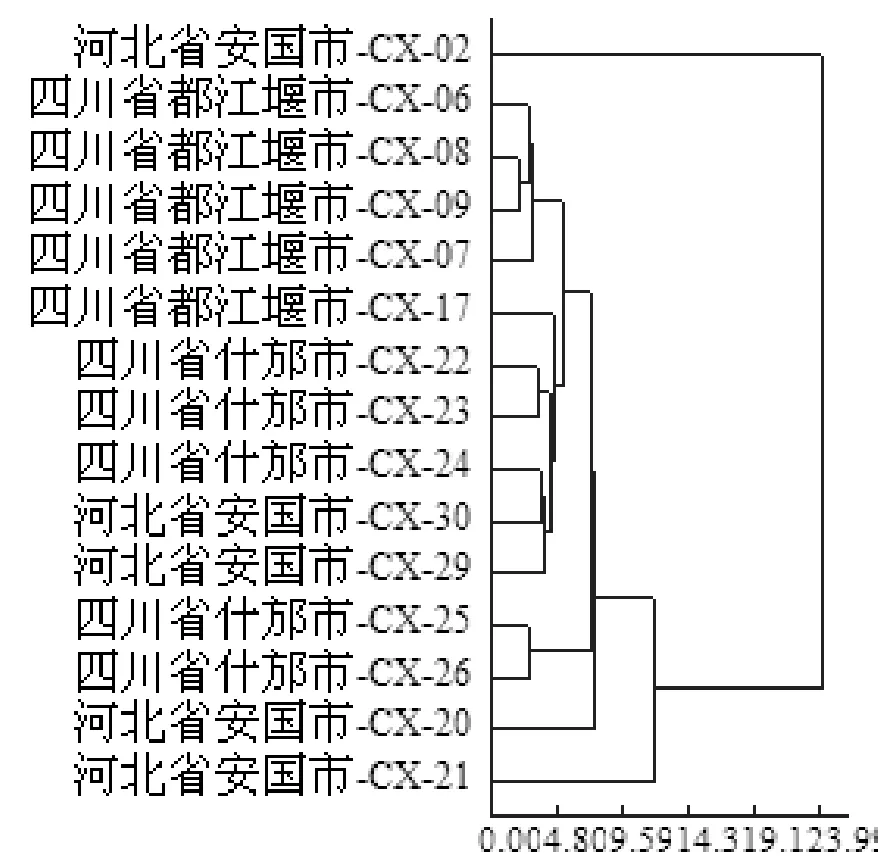
图9 川芎药材指纹图谱聚类分析
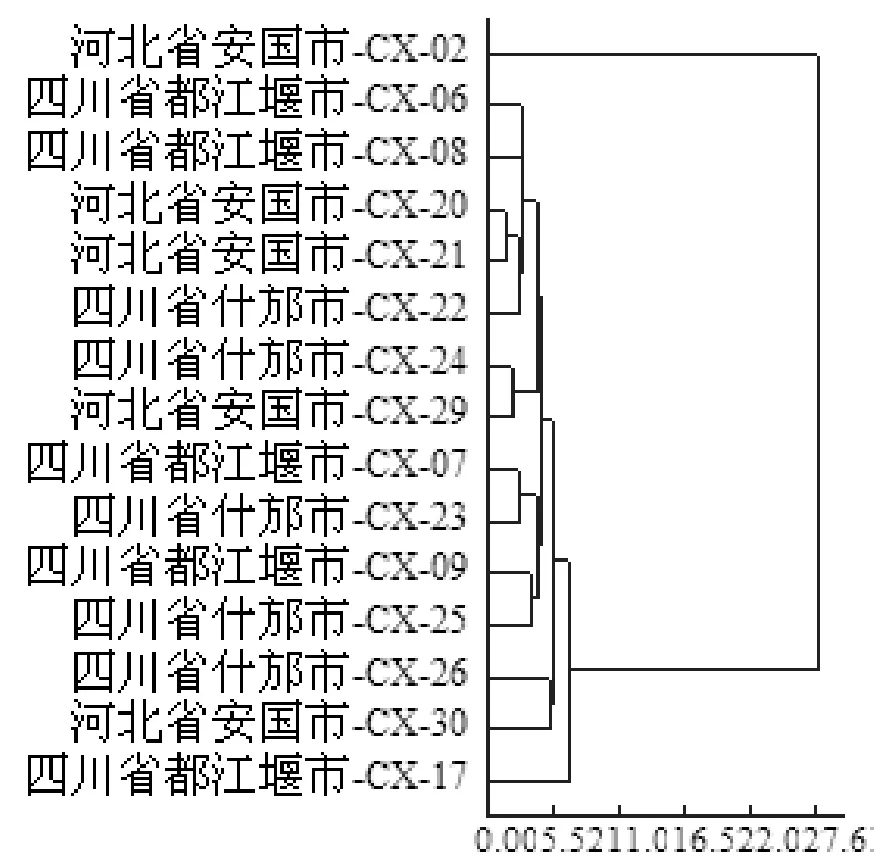
图10 川芎饮片指纹图谱聚类分析
由图9、图10可知,川芎药材及饮片相似度与产地并无太大关联,说明产地来源并不对指纹图谱相似度有显著影响。
2.5 川芎药材和川芎饮片指纹图谱特征分析与比较
对15批川芎药材和川芎饮片指纹图谱相似度进行配对t检验,得配对样本相关系数γ=0.918 8,其均值差异检验t=1.896,df=14,P=0.078 8,结果表明川芎药材和川芎饮片指纹图谱大体一致,川芎炮制前后其化学成分变化无统计学差异(P>0.05)。
3 结论与讨论
川芎药材炮制成饮片过程中需要清洗、闷润(软化)、切片、干燥,川芎中的阿魏酸及挥发油在清洗、干燥中易损失,故川芎饮片指纹图谱可反映川芎炮制前后各成分的变化,从而评价川芎药材-饮片的量值传递率,控制川芎饮片质量。
本研究生成了川芎药材及饮片的对照特征图谱,比较了各批次相似度,发现各批次产地与相似度并无太大关系。
川芎指纹图谱研究多用于不同产地、采收期、川芎饮片及其煎剂、饮片等级[11],以及提取方法[12]等方面差异的比较,而川芎炮制前后质量属性(主要是化学成分)、质量传递的比较(以相似度为评价指标)未见报道,本研究可为川芎产地粗加工、炮制前后质量属性的变化及川芎饮片质量控制提供参考。
