High Genetic Variation of the NorthAmerican Rhus GallAphid,Melaphis Rhois,Compared to its EasternAsian Counterpart,Schlechtendalia Chinensis
2021-12-25RENZhumeiHUANGLanHARRISAJWENJun
REN Zhumei,HUANG Lan,HARRISAJ,WEN Jun
(1.School of Life Science,Shanxi University,Taiyuan 030006,China;2.Key Laboratory of Plant Resources Conservation and Sustainable Utilization,South China Botanical Garden,Chinese Academy of Science,Guangzhou 510650,China;3.Department of Botany,National Museum of Natural History,Smithsonian Institution,Washington,DC 20013,U.S.A)
Abstract:We compared the population genetic diversity and structure of the Rhus gall aphids,Melaphis rhois from North America and Schlechtendalia chinensis from China,by analyzing a dataset of mitochondrial DNA comprising the genes COI,COII,and Cytb representing 32 individuals sampled across three populations of M.rhois and 44 individuals across five populations of S.chinensis.In total,the aligned dataset of mitochondrial sequences was 1 522 bp in length.We found that Melaphis rhois showed considerable variation at 165 nucleotide sites(about 10.6%)of which 155 were parsimony-informative.This is in contrast to S.chinensis,which had only 25 variable sites(about 1.6%)including 20 that were parsimony-informative.Within M.rhois,there were 11 different haplotypes,each of which was found in only a single population except for one that was shared by one individual from Arkansas and five from New Jersey.Within S.chinensis,there were 13 haplotypes,three of which were shared among populations and ten of which were private haplotypes.M.rhois exhibited strong differentiation(FST=0.726 8)according to an AMOVA analysis with the greatest difference between a population from Ohio and all others.The differentiation among populations of S.chinensis was lower(FST=0.237 7).The AMOVA results were corroborated by an analysis with Bayesian analysis of population structure(BAPS),a sta‐tistical parsimony network(TCS),and a reconstructed phylogenetic tree.The high degree of differentiation among populations of M.rhois may result from population subdivision or limited gene flow due to its geographic isolation in North America,separate from all other Rhus-gall aphids(12 of 13 species),which occur in eastern Asia.In contrast,S.chinensis may exhibit less differentia‐tion due to the closer Euclidean distances between populations,anthropogenic effects from cultivation of the galls for traditional medicine,and occasional long distance dispersal of the aphids via water and/or wind.The population of M.rhois in Ohio is unique and genetically distant from other populations,thus it merits additional field work,morphological observations,and expanding the density of within-population sampling to test its taxonomic identity.
Key words:Rhus gall aphids;mitochondrial gene;genetic diversity;comparison
0 Introduction
Rhusgallaphids belong to the subtribe Melaphidina(Hemiptera:Aphididae:Eriosomatinae:Fordini)[1]and show a classic pattern of disjunct distri‐bution in eastern Asia and North America[2-3].This subtribe includes six genera and 12 species,among whichSchlechtendaliachinensisBell is relatively the most common and widely distributed species in the mountain area of Central to Southern China,while on‐ly one genusMelaphisoccurs in North America,andM.rhoisFitch(Hemiptera:Aphididae)was consid‐ered as the only North American representative of the aphid subtribe Melaphidina[4-5].Melaphidina aphids possess a complex life history with cyclic partheno‐genesis and multiple generations co-occurring on the primary host,RhussubgenusRhus(sumac),and the secondary moss hosts.These aphids form galls on the leaves of the primary host plants as part of their life cycle[4,6],and these galls,which are rich in tannins,are known in China as woo-pei-tsze,which is used in medicine and other products,such as dyes[4,7-8].The galls ofS.chinensishave horn-like protrusions and are the most widely cultivated within China[9],while the knobby galls ofM.rhoisare not known to have ever been in cultivation in NorthAmerica.
TheRhusgall aphids from China and from North American were originally regarded as the same species,which has been treated under several differ‐ent names[10].Until 1976,Eastop&Hille Ris Lam‐bers recognized them as separate genera,S.andMelaphis,with species from China and North Ameri‐ca assigned toS.chinensisandM.rhois,respective‐ly[11].More recently,the two genera have been re‐solved as phylogenetically distant within Melaphidina based on both nuclear and mitochondrial DNA da‐ta[12].However,S.chinensisandM.rhoishave rare‐ly been studied comparatively at the genetic level to understand their differences in their genetic diversity and,thus in their population demographics and diver‐sification dynamics.Prior studies of the two species independently have shown low genetic diversity and restricted gene flow inM.rhoisbased on partial DNA sequences and comparatively abundant gene flow among the populations ofS.chinensisaccording to ISSR marker[13].Differences between the two spe‐cies in terms of genetic diversity may arise due to cul‐tivation ofS.chinensisand lack of cultivation inM.rhoisand/orM.rhoishaving two possible sumac hosts,RhusglabraandR.typhina,whileS.chinensisonly has one,R.chinensis.
In this study,we compared genetic diversity of populations ofM.rhoisandS.chinensisusing se‐quences of three protein-coding genes of mitochondri‐al DNA as markers.Our work represents a contribu‐tion toward a fuller assessment of the genetic diversi‐ty within the intercontinental disjunct subtribe and the understanding of the patterns of diversification of these related,disjunct lineages with similarly wide distributions in mesophytic forest habitats in North America and eastern Asia.
1 Material and Methods
1.1 Taxon sampling
We collected samples ofM.rhoisandS.chinen‐sisin USA and China,respectively.All the samples were from natural populations and represented the main distributional areas in North America and east‐ern Asia.Our sampling comprised 79 individuals rep‐resenting 32 individuals ofM.rhoisfrom three popu‐lations,44 individuals ofS.chinensisfrom five popu‐lations,and three accessions of other Melaphidina to represent an outgroup in phylogenetic analyses:Ka‐buragiarhusicola,K.rhusicolaovogallis,andK.rhu‐sicolaensigallis.We collected each sampled individu‐al from different galls on different sumac trees to avoid clonally produced individuals that may occur with the same galls.From each gall,we sampled one aphid individual for genomic DNA extraction for anal‐yses of genetic diversity and stored the remaining aphids in ethanol as voucher specimens at the School of Life Science of Shanxi University,Taiyuan,China.Detailed information about our sampling is presented in Table 1.

Table 1 Sample information for populations of Melaphis rhois and Schlechtendalia chinensis used in this study
1.2 DNA extraction,amplification,and sequenc‐ing
We extracted the total genomic DNA from each aphid individual using a DNA extraction kit(Aidlab Biotechnologies Co.,Ltd)according to the manufac‐turer's protocols.From the total DNA,we performed PCR amplifications of three mitochondrial gene re‐gions(COI,COIIandCytb)using primer sequences from the review article[14].The amplification reactions were at a volume of 50 μL containing 2.5 mmol·L-1Mg2+,0.25 mmol·L-1dNTP,0.4 μmol·L-1primer,1.5 unitTaqDNA polymerase(Bioline Taq),and 20-50 ng template DNA.We ran the PCR cycles using the following program:at 94℃for 3 min,40 cycles at 94℃for 40 sec,at 48℃(except 50℃forCytb)for 30 sec,and 72℃for 1 min,and a final extension at 72℃ for 7 min.The resulting PCR products were se‐quenced in both directions by Shanghai Majorbio Cor‐poration using the same primers as for amplification in an Applied Biosystems ABI 3730 XL analyzer.We assembled the sequences from both directions in Se‐quencer 4.5[15]and aligned them using MUSCLE[16]fol‐lowed by manual adjustment.
1.3 Genetic diversity and phylogenetic analysis
We calculated nucleotide variation of different haplotypes and computed genetic distances between populations in MEGA 5.0 based on pairwise differ‐ences[17].We estimated the haplotype diversity index(Hd),the nucleotide diversity index(π),Fu'sFs[18],and Tajima'sD[19]in DnaSP version 5.10.01[20].For Fu'sFsand Tajima'sD,we generated confidence inter‐vals using 10 000 replicates of a coalescent model with no recombination.We also used TCS 1.21[21]to construct a statistical parsimony network of relation‐ships among different haplotypes.
We performed an AMOVA(analysis of molecu‐lar variance)in Arlequin version 3.5.2[22]to assess the partitioning of molecular variance.We also calculat‐ed pairwise genetic differences with 1 000 permuta‐tions to assess genetic variation between and within populations forM.rhoisandS.chinensis,interped‐ently.
To infer the population structures of each ofM.rhoisandS.chinensis,we performed an untrained clustering analysis in the software package,Bayesian Analysis of Population Structure(BAPS).For this analysis,we concatenated all sequenced mtDNA genes and applied a maximum number of clustersKof 10.
The concatenated dataset from all the haplotypes of the three genes was analyzed by jModeltest version 2.1.7[23]to select the most appropriate evolutionary model using the AIC criterion.We selected the top scoring resulting model that can be set within MrBayes v.3.2.5[24],which we used to perform Bayesian inference(BI).We conducted two indepen‐dent,simultaneous runs of the Markov Chain Monte Carlo(MCMC)for 10 000 000 generations starting from different random trees.We utilized three hot chains and one cold chain for each independent,si‐multaneous run,and sampled the cold chain every 1 000 generations.For each run,we performed burnins in MrBayes of 2 500 trees,and used the remaining trees to construct a 50% majority-rule consensus tree to show posterior probabilities(PP)of clades.We vi‐sualized the consensus tree in Fig Tree.We also con‐ducted maximum likelihood(ML)analyses using RAxML v.8.2[25]with gene partitioning.We selected the GTR model with GAMMA of rate-heterogeneity,and performed bootstrapping in RAxML(ML-BS)with a random number seed and 1 000 replicates.

Fig.1 Assignment of individuals to genetic clusters using BAPS,based mtDNA sequences.Different colors show the differentiation of the M.rhois and S.chinensis populations(C)
2 Results
2.1 Sequence variation
We obtained partial sequences ofCOI,COIIandCytbfor 79 individuals including the outgroups.The combined dataset contained 1 522 characters with 683 bp forCOI,406 bp forCOIIand 433 bp forCytbin length.Nucleotide composition for the three mito‐chondrial genes showed an A+T bias,and the aver‐age A+T base composition among genes ranged from 72.7% to 77.5%.The dataset of the three com‐bined gene sequences showed considerable variability inM.rhois,which had 165 variable nucleotide sites(about 10.6%),and 155 parsimony-informative sites among the individual samples for the species.In con‐trast,variability was lower inS.chinensis,which had only 25 variable sites(about 1.6%),of which 20 were parsimony-informative.
2.2 Genetic diversity
We detected 11 haplotypes representing 32 indi‐viduals from three populations ofM.rhois(Table 2).Most haplotypes were limited to a single population except H2,which we recovered from one individual in Arkansas and five individuals in New Jersey.Hap‐lotypes H1 and H4 were common in Arkansas and New Jersey populations,respectively.M.rhoisex‐hibited a haplotype diversity index(Hd)and nucleo‐tide diversity index(π)of 0.869 0 and 0.035 4,re‐spectively.Among populations ofM.rhois,the Hd and π indices were the highest in the New Jersey pop‐ulation and lowest in the Arkansas population(Ta‐ble 2).
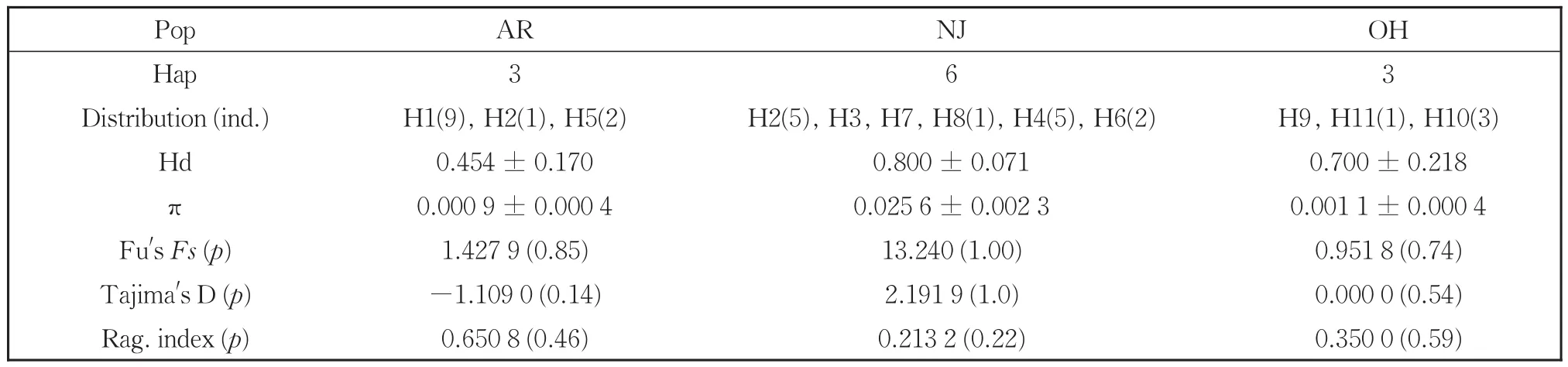
Table 2 Haplotypes,diversity indices,mismatch distribution parameters and neutrality tests from combined mtDNA sequences of M.rhois
InS.chinensis,we detected 13 haplotypes among 44 sequences,most of which were shared among populations.Haplotype H9 was the most com‐mon haplotype and present in 34.1% of the measured samples(Table 3).S.chinensisexhibited a haplotype diversity index(Hd)and nucleotide diversity index(π)of 0.860 5 and 0.003 7,respectively.Among populations ofS.chinensis,the Hd and π indices were the highest in the Guizhou population and low‐est in the Sichuan population.
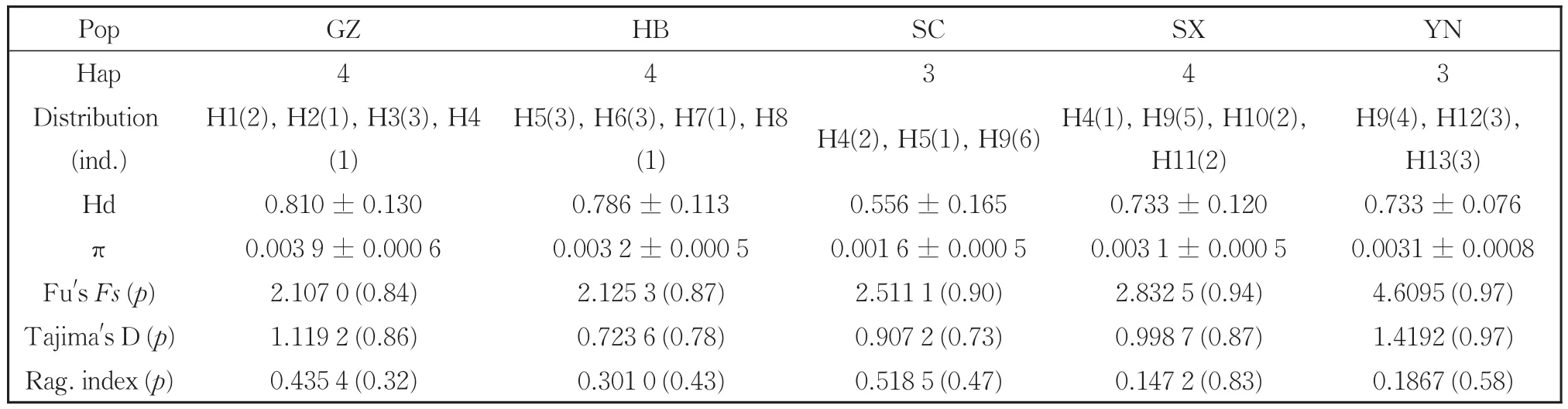
Table 3 Haplotypes,diversity indices,mismatch distribution parameters and neutrality tests from combined mt DNA sequences of S.chinensis.
2.3 Genetic structure and variation
The AMOVA analysis indicated strong popula‐tion differentiation ofM.rhoiswith a fixation index(FST)of 0.726 8(Table 4).A total of 72.68% of the genetic variation was at the population level com‐pared to 27.32% within populations.Populations ofS.chinensisshowed less differentiation with anFSTvalue of 0.237 7,and in contrast toM.rhois,76.23% of the variation was within populations com‐pared to 23.77% among them.We also performed neutrality tests of each population using Fu'sFs.TheFsvalues for all the eight populations of the two spe‐cies were non-significantly positive(Tables 2,3).
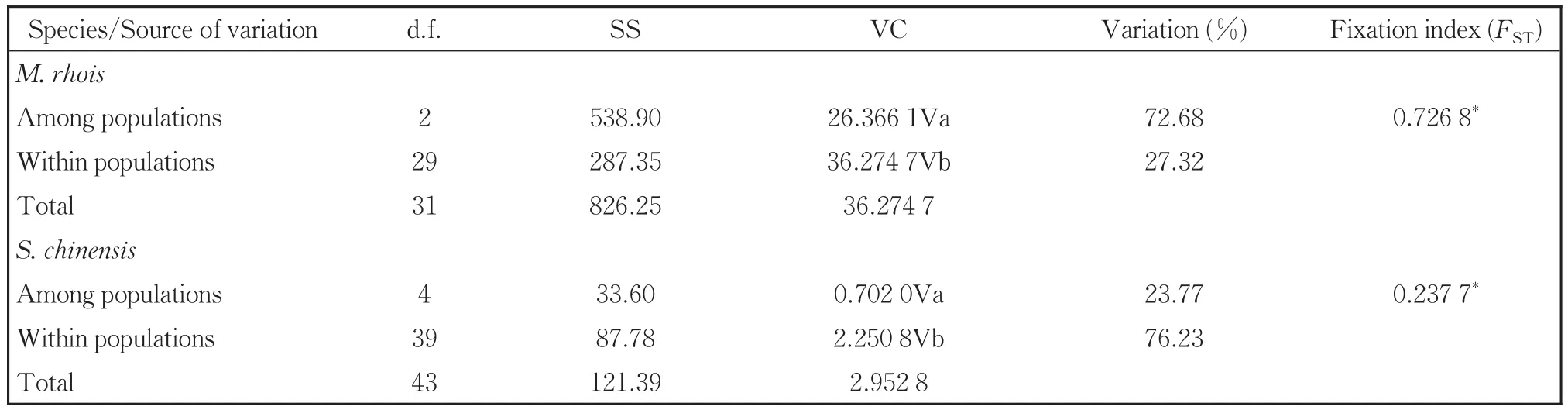
Table 4 Summary of AMOVA results for M.rhois and S.chinensis based on mt DNA sequence data
The BAPS analysis showed that theM.rhoispopulations had a low level of admixture for all the three clusters,whereas there was a higher proportion of admixture inS.chinensispopulation assignment.
2.4 Cluster and phylogenetic analysis
Our phylogenetic analyses of the haplotypes fromS.chinensisandM.rhoisusing BI and ML(Fig.2A)yielded similar topologies with strong sup‐port for most nodes.The 11 haplotypes from the three populations ofM.rhoisformed three clades with high support.
The haplotypes from the Ohio population formed Clade I with high support(ML-BS=100%,PP=1.0),and the Clade II was comprised of the haplo‐types from the New Jersey population(ML-BS=95%,PP=1.0).Clade III was composed of all the haplotypes from the Arkansas population and two hap‐lotypes from the New Jersey population(ML-BS=96%,PP=0.99).Clades III and II are sisters(MLBS=70%,PP=0.96),while Clade I(Ohio popula‐tion)is more distinct from the other two populations.Of the populations ofS.chinensis,all the haplotypes comprised a clade with high support(ML-BS=100%,PP=1.00).However,within the clade,most branches showed low support,so there were no obvi‐ous subclades.
The TCS network of haplotypes showed that hap‐lotype H1 from the Arkansas population ofM.rhoiswas most common(Fig.2B).The population ofM.rhoisfrom Ohio was highly distinct and could not be connected with the rest of the network,largely consis‐tent to the results of the phylogenetic analysis includ‐ing only samples from this study(Fig.2A).Among the populations ofS.chinensis,haplotype H9 was most common and represented individuals from popu‐lations in Sichuan,Yunnan,and Shaanxi.Haplotype H5 was centrally located in the network and,thus,may be ancestral among the populations and individu‐als sampled(Fig.2B).
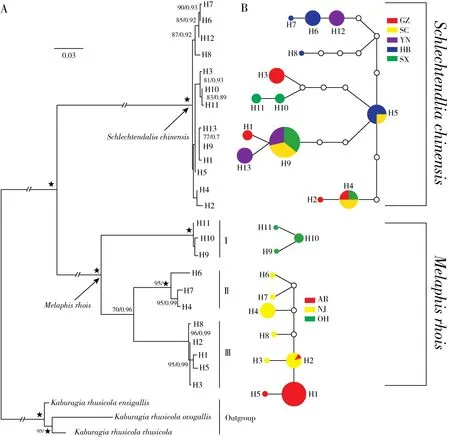
Fig.2 ML tree(A)and TCS network(B)of all haplotypes from M.rhois and S.chinensis populations based on the combined dataset of three mitochondrial genes.Bootstrap values for maximum likelihood(ML-BS>70%)and Bayesian posterior probabilities(BI-PP> 0.90)are shown on the ML tree as ML-BS/BI-PP.Each circle represents a haplotype with a size proportional to its frequency in the sample;the same color represents the same population;white circles depict missing intermediate haplotypes that are inferred by not found among the analyzed individuals,and each line between haplotypes represents a mutation step
3 Discussion
It has been considered that all the species in the aphid subtribe Melaphidina distributed in East Asia with only one speciesM.rhoisFitch(Hemiptera:Aphididae)in North American[5].However,a recent paper suggested that several different genetic clusters ofM.rhoisexist within North America that may con‐stitute different species includingM.asafitchi[26].This is despite our observations that these putative species appear highly similar,so we used the abun‐dantCOIsequences forM.rhoiandM.asafitchiwithin GenBank combined ourCOIdata to verify the identities of the samples ofM.rhoisusing phyloge‐netic analysis.Our phylogeny showed many areas of low support likely due to overall limited variation among sequences.However,we detected two large clades,one comprising largelyM.rhoisand one ofM.asafitchi,and the samples from Ohio are deeply nested within theM.rhoisclade comprise the longest branches within the tree,while samples from New Jer‐sey occurred in both theM.rhoisandM.asafitchiclades.Taken together these data suggest a high level of genetic variation withinMelaphissuch as we inves‐tigate with our other analyses,but suggest that there may be only one hyper diverse species within North America.Based on these findings,we treated all of our samples as belonging toM.rhois,though we ac‐knowledge that greater efforts to sample and under‐stand diversity are needed for this species.
The high haplotype and nucleotide diversity that we observed for populations of the North AmericanM.rhoissuggests that the species might have experi‐enced a long evolutionary history allowing time not only to accumulate differing haplotypes but for con‐siderable differences to arise between them.In con‐trast,S.chinensisshowed high haplotype diversity but low nucleotide diversity,suggesting limited differ‐ences among divergent haplotypes and,thus,a recent population expansion[27].A recent expansion inS.chinensiscould arise from human-mediated move‐ment of genetic material through transplanting of host plants or introduction of aphids in efforts to more widely cultivate the galls and subsequent crossing be‐tween cultivated populations and wild ones[9].
FSTindicates the degree of differentiation among populations,and,following Wright's interpretation ofFSTvalues,0.15-0.25 implies great differentiation and > 0.25 is consistent with very great differentia‐tion[28].We found highFSTvalues(0.726 8)in theM.rhoispopulations and that most of the genetic varia‐tion was among populations.Thus,populations ofM.rhoishave likely undergone considerable differen‐tiation through natural isolation in the wild.InS.chi‐nensis,theFSTvalues were lower and most genetic variation was within populations.Thus,the popula‐tions ofS.chinensislikely experience greater gene flow or have been in more recent contact with one an‐other.The results from BAPS are similar with theFsTto show the highly differentiated populations ofM.rhois(C1,C2 and C3),while more admixture and lower differentiation ofS.chinensispopulations(on‐ly C4).
In bothM.rhoisandS.chinensis,Fu'sFsand Tajima'sDwere not significant,indicating that genet‐ic diversity of the two species has been largely stable over time.This is consistent with the high haplotype and nucleotide diversity inM.rhois,and,taken to‐gether,these values support its long-term evolution‐ary stability.This may be due to long-term relative environmental and geological quiescence in eastern North America[29-31],whereM.rhoisoccurs,com‐pared to recent events,such as mountain-building and changes to the eastern Asian monsoon,affecting Chi‐na[32-33].AlthoughS.chinensismay have undergone or be undergoing a population expansion due to in‐creased demand and cultivation of species,this has yet to leave a sufficiently profound footprint on its ge‐netic history for detection using either Fu'sFsor Taji‐ma'sD.Notably,Fu'sFsand Tajima'sDfor both spe‐cies are non-significantly positive,and positive values indicate a past population bottleneck.Population ge‐netic histories of the species can be more thoroughly explored with additional sampling of populations and individuals using methods that track change through time,such as Bayesian skyline[34].
The phylogenetic trees of samples sequenced for this study and the TCS network(Fig.2)revealed three distinct clades or clusters,respectively,ofM.rhoisand these largely corresponded to population identity and geographic region.No haplotypes in Ohio population were shared by other two popula‐tions,while some individuals from the Arkansas popu‐lation grouped with these from the New Jersey popu‐lation.However,the Arkansas and New Jersey popu‐lations shared only one common haplotype between them.The present results suggest that isolation by distance or early rapid diversification might have played a significant role in the evolution of the spe‐ciesM.rhois[35].The New Jersey population appears to include three divergent haplotypes in three distinct clades within the larger phylogeny.When considered with our network and haplotype phylogeny,this may indicate that the NJ population represent a refugium for diversity in the species,and this can be better ex‐plored with greater sampling from NJ and surround‐ing areas of the northeast Atlantic seaboard of the United States and Canada.
InS.chinensis,some individuals from different populations shared the same haplotypes,but there were no common haplotypes shared among all five populations.However,haplotype H9 was relatively widely distributed and shared by 15 individuals from three populations and may be ancestral among those detected in this study.
Our molecular phylogenetic and network analy‐ses of haplotypes from the three sampled populations revealed that there was greater differentiation among populations of the North AmericanRhusgall aphidM.rhois,compared to its eastern Asian counterpart,S.chinensis.The population from Ohio in particular showed a high level of divergence and could not be connected to the network,and it comprised the lon‐gest branches within the phylogeny of haplotypes and the larger phylogeny including additional sampling,though it is deeply nested within otherM.rhoiswith‐in the larger phylogeny.This suggests that it has un‐dergone considerable diversification following its iso‐lation from the other sampled populations,and the mechanisms driving this diversification can be investi‐gated through additional field work and a more com‐prehensive framework of samples and genetic resourc‐es for populations throughout the North American range.
