Allelic variation in the coumarate 3-hydroxylase gene associated with wood properties of Catalpa fargesii Bur.
2021-12-24NanLuFangMeiZhiWangNanWangYaoXiaoLishengKongGuanzhengQuWenjunMaJunhuiWang
Nan Lu · Fang Mei · Zhi Wang · Nan Wang ·Yao Xiao · Lisheng Kong · Guanzheng Qu ·Wenjun Ma · Junhui Wang
Abstract Coumarate 3-hydroxylase ( C3h) genes participate in the synthesis of lignin and may af fect the properties of wood that are important for its commercial value.A better understanding of the natural variation in C3h genes and their associations to wood properties is required to ef fectively improve wood quality. We used a candidate gene-based association mapping approach to identify CfC3h allelic variants associated with traits that af fect the wood properties of Catalpa fargesii. We f irst isolated the full-length CfC3h cDNA (1825 bp), which was expressed at relatively high levels in xylem according to real timepolymerase chain reaction. In totally, 17 common singlenucleotide polymorphisms (minor allele frequency > 5%)were identif ied through cloning and sequencing the CfC3h locus from a mapping population (including 88 unrelated natural C. fargesii individuals collected from main distribution area). Nucleotide diversity and linkage disequilibrium (LD) in CfC3h indicate that CfC3h has low nucleotide diversity ( π t = 0.0031 and θ w = 0.0103) and relatively low LD (within 1800 bp; r 2 ≥ 0.1). An association analysis identif ied eight common single-nucleotide polymorphisms(SNPs) (false discovery rate, Q < 0.10) and ten haplotypes( Q < 0.10) associated with wood properties, explaining 4.92–12.09% of the phenotypic variance in an association population consisted of 125 unrelated natural individuals (The 88 individuals from the mapping population were comprised in the association population). Our study would provide new insight into C3h gene af fecting wood quality, and the SNP markers identif ied would have potential applications in marker-assisted breeding in the future.
Keywords Catalpa fargesii · Coumarate 3-hydroxylase ·Haplotype-based association analyses · Linkage disequilibrium · Single-nucleotide polymorphisms · Wood properties
Introduction
Trees are important as sustainable and renewable sources of lumber, pulp, and biofuels. Wood formation is actually a major form of carbon sink, involves deposition of secondary cell walls that mainly composed of lignin, hemicellulose,cellulose, and others. The secondary cell wall biosynthesis is a complex and dynamic process that cooperatively regulated by various metabolic pathways involving lignin and polysaccharides according to previous studies (Carocha et al. 2015).
Lignin, a complex racemic mixture of aromatic heteropolymers mainly present in secondary thickened plant cell walls, is the second most abundant organic polymer next to cellulose in trees (Lisperguer et al. 2009). Lignins are essential for plant structure formation and defence. Lignins also support the stem mechanical properties and the cell wall structural integrity (Jones et al. 2001). Downregulation of lignin in poplar signif icantly decreases the elastic modulus,yield stress and modif ies other wood properties (Özparpucu et al. 2017). Wood types vary in their properties, composition and structural or physical features, making wood suitable for dif ferent applications (Du et al. 2013). For example, wood with good mechanical properties, such as stif fness and ultimate stress, is useful as furniture or construction materials. Therefore, forest tree breeding programs should select woods according to the required applications.Catalpa fargesiiBur. (2n = 2X = 40) is a popular timber tree species native to China with straight stem, high density and excellent mechanical characters that makeC. fargesiivaluable materials for the production of furniture and other upmarket woodware (Zhao et al. 2012). Identifying the genes and allelic variations associated with wood quality inC. fargesiiwould yield important information for breeding programs, and will be of practical importance to production (Li et al. 2013). The most important wood traits are complex, quantitative traits,showing phenotypic variation that is typically inf luenced by multiple quantitative trait loci (QTLs) and environmental factors (Resende et al. 2012). Because perennial forest trees have a long lifespan, which make it dif ferent to get an advanced population and usually high heterozygosity, traditional QTL mapping using F1individuals has low mapping resolution such that few alleles are detected (Dillon et al.2012). Indeed, phenotypic variation can sometimes only be detected after years of growth. Linkage disequilibrium (LD)-based association is an ef fective way to examine the associations between natural allelic variation and target traits, and has a higher mapping resolution. Single-nucleotide polymorphism (SNP) markers are usually used in association studies given their wide distribution in the genome and potential to be in LD with the polymorphism (Rafalskia 2002). SNP markers associated with wood properties have been identif ied in several forest trees, such asPopulus(Du et al. 2013;Tian et al. 2014; Wang et al. 2017),Eucalyptus(Thavamanikumar et al. 2014; Resende et al. 2017), and spruce (Lamara et al. 2016; Lenz et al. 2017), among others.
Within the lignin pathway, coumarate 3-hydroxylase(C3h) catalyzes the coumaroyl shikimic acid to caf feoyl shikimic acid, which is a key step to synthesis of guaiacyl and syringyl lignin subunits in dicotyledonous plants (Poovaiah et al. 2014). Down regulation ofC3hreduces lignin content in several plants (Fornalé et al. 2015; Sykes et al.2015). Although functional studies ofC3hhave been carried out, we still know little about their allelic ef fects on the wood properties of trees, which is the foundation for marker-assisted breeding in forest.To study the allelic variation in the coumarate 3-hydroxylase gene and associated with wood properties and we f irst cloned a gene encoding theC3hhomologCfC3hfromC. fargesii, and measured its expression in specif ic tissues. Single-marker and haplotypebased association methods were combined to identify factors underlying natural variations in wood properties in aC.fargesiipopulation. This is the f irst association study about the allelic variations in theC3hand wood property. Molecular markers identif ied in our study would lay a foundation for improving wood quality through molecular breeding ofC. fargesiiin the future.
Materials and methods
Plant materials and DNA extraction
TheC. fargesiipopulation in this study consisted of 125 unrelated individuals growing in the Xiaolongshan conservation area, Gansu Province, China (33°40′ N, 106°23′ E)(Zhao et al. 2012). Branch segments of the 125 individuals were collected from eight cities in four provinces covering the main natural distribution range ofC. fargesiiand grafted to establish a clonal plantation in 2009 using a randomised complete block design with two plants per clone in each block and six replications (“row spacing” is 2 m and “plant spacing” is 2 m). These individuals were divided into four groups by geolocation: Fenhe River valley, Jinghe River valley, Jialingjiang River valley and Yellow River valley. The 88 unrelated individuals were selected from among these groups to identify SNPs associated with wood properties via polymerase chain reaction (PCR) amplif ication and sequencing. (At least one individual was selected for each location).
Fresh leaves were collected from each individual and total genomic DNA was extracted using the DNAeasy Plant kit (Qiagen, Shanghai, China) following the manufacturer’s protocol.
Phenotypic data
Nine phenotypic traits associated with wood property,including wood basic density and eight microstructural characteristics were measured: pore rate, cell wall percentages (the percentage of cell wall in whole cells), cell wall thickness, radial lumen diameter, chordwise lumen diameter,radial f ibre central cavity diameter, chordwise f ibre central cavity diameter, and average f ibre central cavity diameter.The nine traits were selected for their possible inf luences on the f inal mechanical properties of timber according the other studies (Li et al. 2015). The 125 individuals were sampled in 2012. Cores containing bark and pith were collected at breast height (1.3 m above the ground) from stems of trees in the south-facing direction to evaluate wood basic density and other properties using an increment borer (7 mm). Wood basic density was measured as follows (Eq. 1):
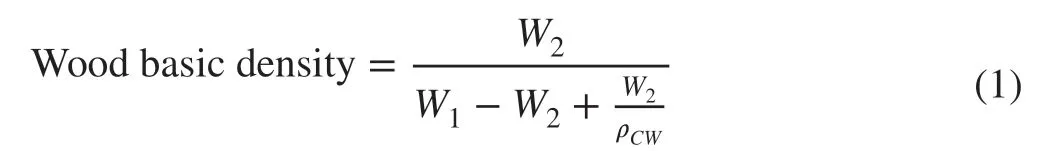
where,W1,W2,andρcwrepresent water-saturated weight,oven dry weight and cell wall density (here we used the constant 1.53 g cm−3for ρcw), respectively (Zheng et al. 2015;Duan et al. 2016).
The anatomical parameters (pore rate, cell wall percentage, cell wall thickness, radial lumen diameter, chordwise lumen diameter, radial f ibre central cavity diameter,chordwise f ibre central cavity diameter and average f ibre central cavity diameter) were determined according to Li et al. ( 2015): the cores were split into 3-cm-long pieces and cross-Sects. (10–15 μm thick) were prepared using a sliding microtome (Leica, Heidelberg, Germany), stained with Safranin-O (1% in distilled water) and permanently f ixed with Eukitt (BiOptica, Milan, Italy) (Li et al. 2015). A digital image processing system, including a light microscope(80i; Nikon, Tokyo, Japan), video camera sensor (Penguin 600CL; Pixera, Santa Clara, CA, USA) and TDY-5.2 colour image analysis system (Beijing Tiandiyu Science and Technology Co., Ltd., Beijing, China) were used to measure the wood microstructural characteristics (Wang et al. 2005).
The frequency distributions for each trait are calculated using Excel (ver. 2013; Microsoft, Redmond, WA, USA)and shown in Fig. S1. The phenotypic data are listed in Table S1. SPSS software (ver. 18.0; SPSS Inc., Chicago,IL, USA) was used to evaluate the nine phenotypic traits,including in terms of mean values, ranges and coef ficients of variation (Table S2). The proportion of the phenotypic variance explained by population structure (Table S2) was evaluated by SAS software (ver. 9.1.3; SAS Institute Inc.,Cary, NC, USA) using generalized linear model (GLM). The variance components and narrow-sense heritability (h2) were evaluated using R/ASReml (ver. 4.0; VSN International Ltd.,Hemel Hempstead, UK).
Isolation of the whole coding sequence (CDS)and genomic DNA amplif ication of the C3h homologue in Catalpa fargesii
Total RNA was extracted from the young branches of a 1-year-old “Xianhuiqiu” (C. fargesii) clone using the Plant Qiagen RNAeasy kit (Qiagen) according to the manufacturer’s instructions. First-strand cDNA was synthesised from 2 g of DNase I-treated RNA using the PrimeScript™ 1st Strand cDNA Synthesis Kit (TaKaRa Bio, Shiga, Japan).The entire open reading frame (ORF) of theC3hhomologue fromC. fargesiiwas isolated in the following way: We f irst obtained the partialC3hhomologue sequence from previous RNA-seq data, i.e. an internal coding region of aC3hhomologue sequence according to the National Center for Biotechnology Information (NCBI) database. The 3′ end was isolated by 3′ rapid amplif ication of cDNA ends (RACE)using the 3′-full RACE Core Set (ver. 2.0; TaKaRa Bio) and designed primers (C3h-3′ RACE; Table S3), and a 3′ RACE adaptor primer (C3h-3′ RACE adaptor primer; Table S3).To isolate the 5′end, 5′ RACE was carried out using the 5′-full RACE Core Set (ver. 2.0; TaKaRa Bio) according to the manufacturer’s instructions using specif ic primers, i.e.a designed 5′RACE primer (C3h-5′ RACE; Table S3) and a 5′ RACE adaptor primer (C3h-5′ RACE adaptor primer;Table S3). PCR was carried out using the C3h-CDS primers to verify the integrity of theC3hhomologue CDS sequence(Table S3).
Total genomic DNA was extracted from young leaves of a 1-year-old “Xianhuiqiu” clone with the DNAeasy Plant Mini kit (Qiagen). The intron sequences were cloned using specif ic primers designed based on the cDNA sequences C3ha, C3h-b and C3h-c, and were then sequenced (Table S3).After PCR amplif ication, three fragments were cloned into pMD 19-T Vector (Takara Bio) and sequenced. The entireCfC3hDNA sequence was obtained according to the assembly result of sequenced fragments using DNAman software(Lynnon BioSoft, Vaudreuil, Quebec, Canada). The entireCfC3hDNA sequence was identif ied using C3h-d primers(Table S3).
Sequence alignment and phylogenetic analyses
TheCfC3hamino acid sequences were subjected to BLAST analysis via the GenBank database ( http://www.ncbi.nlm.nih.gov/sites/entre z?db=nucle otide). During the BLAST searches, multipleC3hproteins from various species were selected for alignment using DNAMan software.To analyse the phylogenetic relationship ofCfC3hto theC3hgenes from other species, the amino acid sequences of theC3hgene fromSesamum indicum(AAL47545.1),Scutellaria baicalensis(BAJ09387.1),Salvia miltiorrhiza(ACA64048.1),Coffea Arabica(AFP49812.1),Populus tomentosa(AFZ78540.1),Platycodon grandifloras(AEM63674.1),Caragana korshinskii(AEV93473.1),Neosinocalamus affinis(AFD29885.1),Panicum virgatum(BAO20879.1),Ginkgo biloba(AAY54293.1),Cunninghamia lanceolate(AFX98060.1),Narcissus tazetta(AGI97941.1),Pinus taeda(AAL47685.1) andIsatis tinctori(AEH20527.1) were downloaded from NCBI ( http://www.ncbi.nlm.nih.gov) and aligned with the ClustalW program using the default settings. The phylogenetic tree was constructed using MEGA 5.0 software. The phylogenetic tree was constructed based on the maximum likelihood method with the following parameters: bootstrap (1000 replicates),Jones–Taylor–Thornton substitution model, uniform rates,partial deletion of gaps/missing data and nearest neighbour interchange.
Expression of CfC3h in dif ferent C. fargesii organs
We collected tissue and organ samples of tree bark, phloem,xylem, leaves, f lowers and juvenile branch meristem from three 11-year-old Xianhuiqiu (C. fargesii) trees planted in Luoyang, Henan in the middle of April for RNA extraction.Each tree was considered as one repetition. All tissues were immediately frozen in liquid nitrogen and stored at − 80 °C.The RNA extraction and cDNA synthesis procedures were performed as described above. Tissue-specif ic expression analysis ofCfC3hwas done using real-time quantitative polymerase chain reaction (RT-qPCR).
RT-qPCR was performed using a LightCycler 480 System (Roche, Basel, Switzerland) and the SYBR Premix Ex Taq Kit (TaKaRa Bio), under the amplif ication conditions recommended by TaKaRa Bio. TheCfC3h-specif ic primers(C3h-q) were designed using Primer Express 5.0 software(Applied Biosystems, Foster City, CA, USA) and the actin gene was selected as the internal control according to Jing et al. ( 2015). The PCR program included initial denaturation at 95 °C for 30 s, and 40 cycles for 5 s at 95 °C and 30 s at 60 °C. All reactions were carried out four times, and the tissue-specif ic expression levels were calculated using the 2−ΔΔCTformula .
SNP identif ication and genotyping
To identify SNPs within theCfC3hgene, the 44 bp 5′-untranslated region (UTR), the entire coding region and the 128 bp 3′ UTR were sequenced and analysed in 88 unrelated individuals from the mapping population, to identify SNPs without consideration of insertions/deletions.To ensure the accuracy of sequencing, three pairs of primers (C3h-1, C3h-2 and C3h-3) were used to amplify three fragments of the entire sequence (Table S3). Primer pairs for amplif ication were designed using Primer Express 5.0 software. DNAMAN and ClustalX2 (Larkin et al. 2007)were used for sequence alignment, and manual editing was performed to conf irm sequence quality. Eight clones of each individual were used to identify putative SNP variants, and fragments were randomly selected for initial allele sequencing via the ABI3730XL instrument (Applied Biosystems).The 88 genomic clones were aligned and compared using MEGA5.0 (Tamura et al. 2011) and DnaSP v5 software (UB Web, Barcelona, Spain, 2010) to identify SNPs and analyse the nucleotide polymorphisms. Common SNPs (SNPs with minor allele frequencies more than 5%) were genotyped across all 125 DNA samples from the overall population.The genotypic data ofCfC3hidentif ied in this population were shown in Table S4.
Nucleotide diversity and linkage disequilibrium analysis
Summary statistics for the SNP polymorphisms were generated by linear regression analysis using DnaSP v5. Nucleotide diversity was estimated according to the average number of pairwise dif ferences per site between sequences,π(Nei 1987), and the average number of segregating sites,θw(Watterson 1975). The HAPLOVIEW software package( http://www.broad.mit.edu/mpg/haplo view.html) was used to assess LD among the common SNPs. The squared allelic correlation coef ficient (r2) was used to estimate LD (Hill and Robertson 1968). The signif icance (Pvalue) ofr2for each SNP locus was calculated using 100,000 permutations.
SNP-based associations and modes of gene action
Single-marker models were created for all SNP–trait combinations. A mixed linear model (MLM) was f itted to each trait-SNP combination using TASSEL v5.0 software. The MLM, from theQ+Kmodel, uses the values of estimated membership probability (Q) to evaluate the ef fects of population structure, and those of pairwise kinship (K) to evaluate relatedness among individuals for marker-trait associations.TheQmatrix was prepared, from the pattern of the population structure (K= 3) within the overall population (125 unrelated individuals), using STRU CTU RE (ver. 2.3.1). TheKmatrix was obtained via the method proposed by Ritland ( 1996) using the SPAGeDi program (ver. 1.2). Corrections for multiple testing were performed using the positive false discovery rate(FDR) method in QVALUE software (Storey and Tibshirani 2003). The percentage of phenotypic variation (R2) explained by each SNP was calculated by using the below formula:

where, SSt and SST represented the variance between genotypes and the total variance, respectively. The detailed information can be found in Lu et al. ( 2018).The ratios of dominant (d) to additive (a) ef fects calculated from least square means for each genotypic class were used to quantify the modes of gene action according to Wegrzyn et al.( 2010). Values of |d/a| in the range of 0.50 to 1.25 were considered to indicate partial or complete dominance, and those in the range of |d/a| no more than 0.5 were considered additive ef fects. In addition, values of |d/a| more than 1.25 were considered to indicate under- or over-dominance. The detailed algorithm and formulas for estimating the gene action were described by Eckert et al. ( 2009).
Haplotype analysis
Within the genotypic data of 125 individuals, we analysed the haplotypes based on information of the contiguous common SNPs. Haplotype frequencies were estimated, and haplotype association tests were performed using a three-marker sliding window via haplotype trend regression software (Zaykin et al. 2002). We used a 1000 permutation tests to evaluate the signif icance of haplotype-based associations and haplotypes with a frequency ≥ 1% were selected for further analysis. The multiple testing was corrected using a positive FDR (Q≤ 0.1)in QVALUE.
Results
Cloning of Catalpa fargesii C3h
The full-lengthCfC3hcDNA isolated using RACE was 1825 bp, including a 1530 bp ORF encoding 510 amino acids and a 69 bp 5′ UTR sequence, as well as 226 bp 3′ UTR sequence. The full-lengthCfC3hDNA sequence was 3511 bp and contained a 3104 bp coding region, f lanked by a 125 bp 5′UTR sequence and a 282 bp 3′ UTR sequence (Fig. 1). Alignment of the cDNA sequence to the full-length DNA sequences showed thatCfC3hhas three exons and two introns.
C3hgenes were divided into four groups according to the molecular phylogeny analysis.CfC3hbelongs to group IV, the same group asC3hgenes of other three Tubif lorae species,namelySesamum indicum,Salvia miltiorrhiza,andScutellaria baicalensis. It is interesting that two dicotyledonous groups(group I and IV) belonged to two dif ferent branches, and the genetic evolutionary relationships of theC3hgenes from group I were closer to genes from monocotyledons and gymnosperms than those from group IV. The phylogeny analysis suggested that separation ofC3hgenes may have occurred before gymnosperms and angiosperms diverged (Fig. 2). The sequence alignment showed thatCfC3hinC. fargesiihad close similarity at the amino acid level toC3hfrom other species (Fig. 3).C3hbelonged to the P450 superfamily, and a cytochrome P450 cysteine heme–iron ligand signature (FGXGRRXCPG) was also found in the C terminal region ofCfC3hfrom F (432) to G (441).
Expression of CfC3h in dif ferent organs
We used RT-qPCR to determine the tissue-specif ic expression ofCfC3hinC. fargesii. As shown in Fig. 4, the expression ofCfC3hwas highest in xylem (0.406 ± 0.048), followed by phloem (0.229 ± 0.056) and leaves (0.188 ± 0.056). It was the lowest in f lowers (0.022 ± 0.006). This result shows thatCfC3his mainly expressed in xylem.
Phenotypic variations in the Catalpa fargesii population
Phenotypic variations in all nine traits were evaluated in the overallC. fargesiipopulation (125 individuals) to test quantitative traits for association mapping. All traits varied signif icantly within the overall population; for example, pore rate,cell wall percentages and radial lumen diameter ranged from 6 to 14% (mean, 9.94%), 22.95% to 41.38% (mean, 35.03%),and 7.31 to 27.74 μm (mean of 14.96 μm), respectively. To estimate the phenotypic variance within the overall population,we computed the coef ficient of variation (CV) for all nine traits(Table S2). The results demonstrated that pore rates had the highest CV (14.45%), followed by cell wall thickness (12.98%)and radial lumen diameter (9.82%). All nine traits followed an approximately normal distribution (Fig. S1).
Nucleotide diversity and linkage disequilibrium in CfC3h

Fig. 1 Genomic organisation of CfC3h

Fig. 2 An unrooted phylogenetic tree of C3h members from dif ferent species. Sesamum indicum (AAL47545.1): SiC3H; Scutellaria baicalensis (BAJ09387.1): SbC3H; Salvia miltiorrhiza (ACA64048.1):SmC3H; Cof fea Arabica (AFP49812.1): CaC3H; Populus tomentosa(AFZ78540.1): PtoC3H; Platycodon grandif lora (AEM63674.1):PgC3H; Caragana korshinskii (AEV93473.1): CkC3H; Neosinocalamus af fi nis (AFD29885.1): NaC3H; Panicum virgatum(BAO20879.1): PvC3H 1; Ginkgo biloba (AAY54293.1): GbC3H;Cunninghamia lanceolate (AFX98060.1): ClC3H; Narcissus tazetta(AGI97941.1): NtC3H; Pinus taeda (AAL47685.1): PtaC3H; Isatis tinctori (AEH20527.1): ItC3H; Catalpa fargesii Bur.: CfC3H
We amplified and sequenced a 3276 bp genomic region ofCfC3hfrom 88 unrelated individuals within the overall population, including the 44 bp 5′ UTR, the entire coding region and the 151 bp 3′ UTR sequence, to determine SNP diversity. Alignment of the 88 samples revealed a total of 163 SNPs inCfC3h, with a polymorphism of 4.94% (Table 1). Of the 163 SNPs, only 17 (10.43%)were considered common SNPs (Fig. S2). The highest level of nucleotide polymorphism in the coding regions occurred in intron 2 (7.22%), and the lowest in exon 2(2.76%). TheCfC3hlocus had low nucleotide diversity,withπ t= 0.0031 andθ w= 0.0103 (Table 1). Specifically,nucleotide diversity ranged from 0.0024 (exon 2) to 0.0094 (5′ UTR), andθ wvaried between 0.0060 (exon 2)and 0.0270 (5′ UTR). The coding region had more nonsynonymous changes (40) than synonymous changes (15).
The SNPs identified in the 88 unrelated individuals were used to calculater2and the LD level was assessed according to the pattern ofr2with base-pair distance within theCfC3h. Ther2value decreased to 0.1 within 1800 bp (Fig. 5), indicating that LD may not extend over the entire region that we sequenced. We then genotyped 17 SNPs common across 125 individuals, and LD analysis using genotype data revealed four distinct haplotype blocks within theCfC3hlocus: from SNP 6 to 7, 9 to 10,11 to 12 and 14 to 15 (Fig. 6). LD between the SNPs was relatively high within each block (r2> 0.75).
SNP-trait associations
MLM was used to detect associations between phenotypes and genotypes for the dif ferent SNPs, after correcting for multiple testing using the FDR method (Q≤ 0.1). We identif ied eight signif icant associations at a threshold ofP≤ 0.05, encompassing seven unique SNPs (SNP 1, SNP 2, SNP 3, SNP 5, SNP 9, SNP 10 and SNP 17) signif icantly associated with wood basic density, pore rate, cell wall percentage, cell wall thickness and chordwise lumen diameter (Table 2), explaining 4.92−7.99% of the phenotypic variance in these traits. Five of these eight associations were consistent with over-dominance modes of gene action (|d/a|> 1.25) and one association was partial or complete dominance (Table 3). Of the seven signif icant SNPs, f ive were located in exons, including four nonsynonymous and one synonymous SNP. The nonsynonymous marker, SNP 1, showed an amino acid change from Val to Leu in exon 1, and was signif icantly associated with cell wall thickness, explaining 6.85% of the phenotypic variance therein. Heterozygous trees (CG) exhibited higher cell wall thickness (2.93 μm) than trees with the CC and GG genotypes (2.83 and 2.82 μm, respectively). The SNP 5 was signif icantly associated with pore rate, explaining 4.92% of the variance and exhibiting over-dominance for this trait (|d/a|> 1.25). The GG genotype of SNP 3 exhibited a lower cell wall percentage (32.61%) compared with the CG and CC genotypes (36.58% and 35.15%, respectively) (Fig. S3), thus exerting an over-dominance ef fect on cell wall percentage (|d/a|> 1.25). In the 3′ UTR, SNP 17 was associated with wood basic density (explaining 6.08% of the variance therein) and the mean values of two main genotypic groups: TT and TC were 0.417 and 0.429 g cm−3, respectively. SNP 10 was significantly associated with wood basic density (explaining 7.99%of the variance therein) and chordwise lumen diameter(6.39% of the variance).

Fig. 3 Sequence comparison of CfC3h with other C3h proteins. The sequence of the cytochrome P450 cysteine heme-iron ligand signature is shown by the red box. Sesamum indicum (AAL47545.1): SiC3H;Scutellaria baicalensis (BAJ09387.1): SbC3H; Salvia miltiorrhiza(ACA64048.1): SmC3H; CaC3H; Populus tomentosa (AFZ78540.1):PtoC3H; Platycodon grandif lora (AEM63674.1): PgC3H; Caragana korshinskii (AEV93473.1): CkC3H; Catalpa fargesii Bur.: CfC3H
Haplotype-based association tests were performed to identify haplotypes signif icantly associated with the nine phenotypic traits (Table 4). This analysis identif ied 10 associations between 11 common haplotypes (frequency ≥ 1%) in six blocks. Eight of the traits (i.e. all except average central diameter) reached the signif icance threshold ofP≤ 0.05 and FDR ≤ 0.1 among the entire region. Of these, three haplotypes from SNPs 5–7 were associated with pore rate, cell wall percentage and cell wall thickness, and three haplotypes from SNPs. 15–17 were associated with wood basic density,radial lumen diameter and radial f ibre central cavity diameter. The proportion of phenotypic variation explained by these haplotypes ranged from 6.32 to 12.30%.
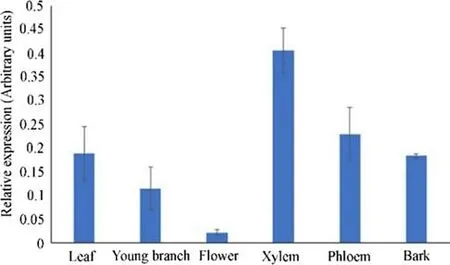
Fig. 4 Levels of the CfC3h transcript in dif ferent organs. Error bars represent the standard deviation of three biological replicates
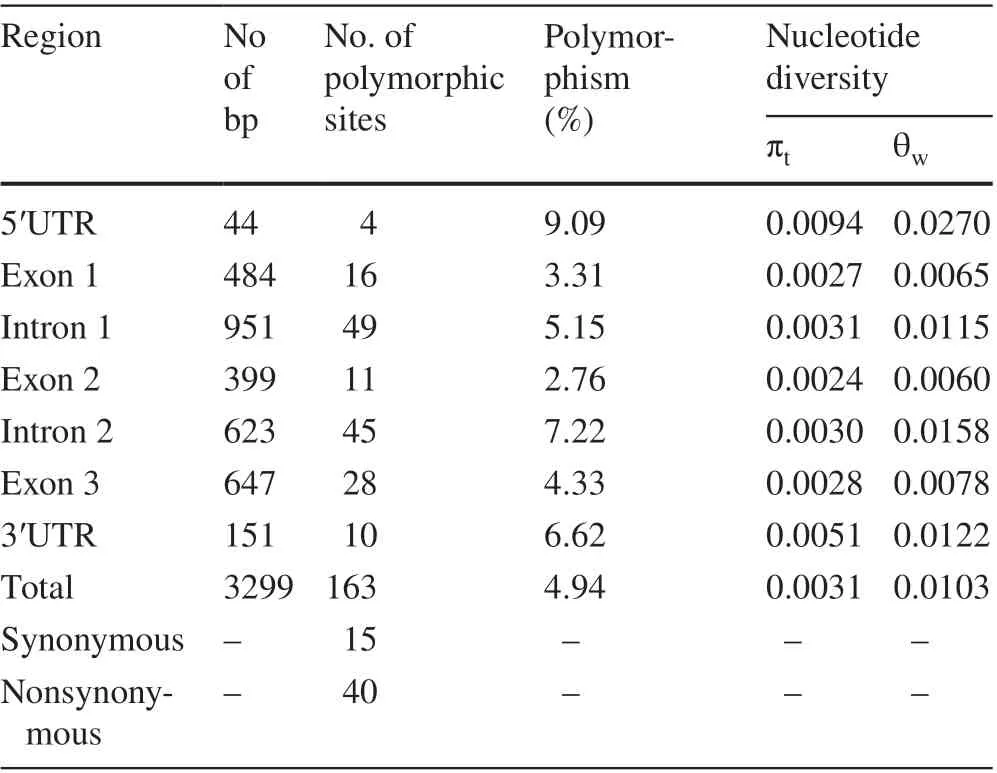
Table 1 Nucleotide polymorphisms in the CfC3h locus of Catalpa fargesii
Discussion
The putative function of CfC3h
C3his an important enzyme in lignin synthesis, where lignins are a major component of plant secondary cell walls.C3hmutants have been studied inArabidopsis thalianain the context of recovery of the function of theC3hgene (Kim et al. 2014). Defects in coumarate 3′-hydroxylase cause dwarf ism and reduce cell wall lignin content. Wang et al.( 2018) reported that downregulation ofC3hin poplar not only reduces lignin levels, but also markedly increase the proportion of G and S-type lignin, and f inally inf luence the wood properties.
We cloned aCfC3hgene fromC. fargesii, which shared 69% and 71% identity at the nucleotide level withArabidopsis C3h(AT2G40890) andPopulus alba×grandidentata C3h(GenBank accession no. EU391631), respectively. We further analysed the expression ofCfC3hin dif ferent organs and observed the highest expression in xylem, which may due to the higher degree of lignif ication in xylem.
This study identif ied an association between allelic variation inCfC3hand several wood quality traits, including cell wall percentage and cell wall thickness (Table 2). These results are consistent with previous studies and conf irmed the importance ofC3hwithin the structure of the secondary cell wall (Ralph et al. 2006; Fornalé et al. 2015).

Fig. 5 Decay of LD within CfC3h based on sequences of the CfC3h region from 88 unrelated individuals. We sequenced the CfC3h regions of 88 unrelated individuals. Pairwise correlations between singlenucleotide polymorphisms(SNPs) are plotted against the physical distance between the SNPs in base pairs. The curves show thelinear regressionof r2 accordingto the physical distance in the base pair

Fig. 6 Four distinct haplotype blocks within the CfC3h gene.The percentage (%) of pairwise LD ( r 2 ) is shown by the numbers in the coloured squares.Dashed lines indicate the physical locations of the SNPs within the gene
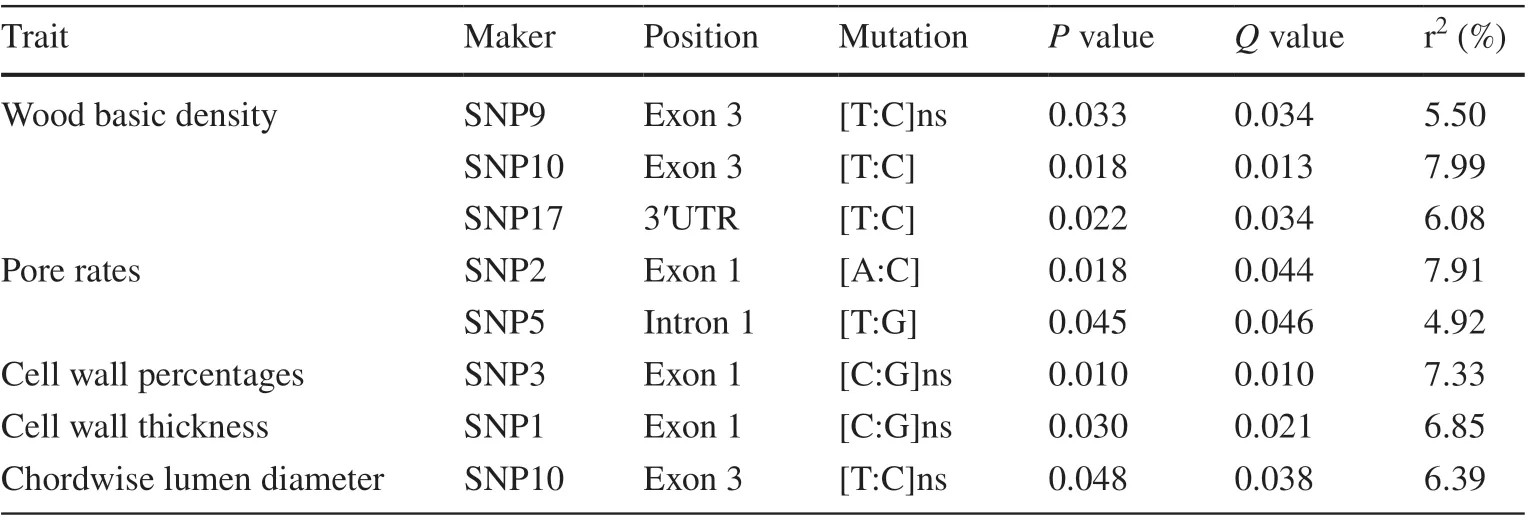
Table 2 Single-nucleotide polymorphism markers signif icantly associated with wood traits in the overall Catalpa fargesii population(n = 125)

Table 3 The modes of gene action for signif icant marker–trait pairs

Table 4 Haplotypes signif icantly associated with the wood traits
Nucleotide diversity and LD in CfC3h
An understand of the extent of LD and nucleotide diversity level in a natural population could evaluate the precision and ef fectiveness of association mapping, as well as ref lecting the forces in charge of the evolutionary change (Zhang et al.2010). So, a comprehensive study of the patterns of SNP distribution and frequency within theCfC3hlocus of from theC. fargesiipopulation is necessary before SNP-based association mapping. The SNP frequency in exons regions,intron regions and the genomic sequence was 3.59%, 5.97%,and 6.62%, respectively. The exons showed substantially lower levels of nucleotide diversity compared with introns in the coding region (Table 1), which is consistent with previous studies (Du et al. 2013; Wang et al. 2017) and indicates that the exon regions may have undergone strong purifying selection and thus remained relatively conserved.The sequence that codes the cytochrome P450 conserved domain (FGXGRRXCPG) was located in exon 3, which had a low level of nucleotide diversity (πt= 0.0027) thus indicating thatCfC3his extremely conserved due to its crucial role in the synthesis of monolignols and other 3,4-hydroxylated phenylpropanoid secondary metabolites (Bate et al.1998; Kim et al. 1998). Compared to our previous study,the nucleotide diversity ofCfC3hwas similar to that ofCfSUS(πt= 0.0031) (Lu et al. 2018), which indicated that the two genes may have a similar pattern of genetic variance in the natural population. However, and nucleotide diversity detected in a population may inf luenced by the population size, sampling strategy and other factors (Tian et al. 2014),so in the future study, larger population and more reasonable sampling strategy should be used to evaluate the nucleotide diversity level ofC. fargesii.
Understanding the level of LD can help to determine whether candidate gene-based association studies are appropriate for understanding the molecular basis underlying quantitative variation, and whether a genome-wide approach is feasible (Du et al. 2013). In our study,CfC3hshowed a relatively low level of LD and a rapid decline, indicating that candidate gene-based association studies may be appropriate in this instance to identify SNPs responsible for the detected traits. In fact, a low and rapidly declining LD has been reported in other studies (Guerra et al. 2013; Chu et al. 2014), which may due to the outcrossing habit, long history of recombination and large population size of these species (Abdurakhmonov and Abdukarimov 2008). The LD level ofCfC3hwas similar to that ofCfSUS(r2< 0.1, within 1600 bp) in the same population (Lu et al. 2018). Additionally, we detected four distinct haplotype blocks within theCfC3hgene and the distances between adjacent SNPs in the blocks were small (20 to 79 bp). Low LD observed inCfC3hgene may suggested high resolution of marker-trait associations.
Determining the allelic polymorphisms underlying wood properties
In our study, a gene-based association analysis has been used to identify alleles associated with wood properties in several tree species includingPopulus,Eucalyptusand somePinusplants. However, SNP association studies have not been reported forC. fargesii. Therefore, we employed single-marker and haplotype-based association studies of a candidate gene inC. fargesii.The results showed that several single SNP markers and haplotypes were associated with wood properties in ourC. fargesiipopulation, indicating that these markers may be in close proximity to, or in fact are,the functional variant.
The eight single-SNP associations identif ied in our study only explain a small proportion of the variance in wood traits, which is in accordance with previous studies of other tree species (Porth et al. 2013; Wang et al. 2017). This may be because wood traits are usually quantitatively characterised and controlled by multiple genes. In addition, most of the SNPs (f ive of seven) were located in exon regions and the mode of gene action of SNP1 and SNP3 was overdomination. Mutations in coding regions, particularly nonsynonymous mutations, can af fect gene function. Vanholme et al. ( 2013) identif ied a stop codon mutant in the hydroxycinnamoyl-CoA: shikimate hydroxycinnamoyl transferase gene, resulting in modif ied lignin composition inPopulus nigra. The four SNPs (SNP1, SNP3, SNP9 and SNP10) were located in exons and identif ied as nonsynonymous mutations. It remains unclear how amino acid changes in these four locations inf luenced the function ofCfC3h. However,Wang et al. reported that transfer between amino acids with similar polarities, charges or sizes, such as Cys and Ser, may not af fect the function of genes (Wang et al. 2017). Whether amino acid changes of these four locations inf luence the function ofCfC3hgene, need to be further studied.
Wood basic density is one of the most important factors associated with wood mechanical strength. Our study showed that SNPs 9 and 10 explained 5.50% and 7.99% of the variance in wood basic density, while a haplotype from SNPs 9 to 11 explained 11.59% of the phenotypic variation, slightly higher than single-marker association (5.50%and 7.99%), indicating that markers surrounding SNPs 9 and 10 may interact with the two loci and contribute to phenotypic ef fects; however it need be further investigated.SNP 5 (located in intron 1) was associated with 4.92% of the variation in pore rate, which was lower than that of a haplotype from SNPs 5 to 7 (8.51%). SNP 5 may interact with loci nearby or loci that inf luence RNA splicing, and thus inf luence the pore rate; however, further investigation is required to reveal the detailed mechanisms. Notably, SNP 17 in the 3′ UTR region was signif icantly associated with wood basic density. Although the polymorphism in the 3′UTR region did not alter the amino acids, 3′ UTRs participate in the regulation of gene expression by af fecting mRNA deadenylation and degradation (Fang et al. 2010).In addition, SNPs in 5′ UTR region can af fect gene regulation by inf luencing transcriptional binding (Beaulieu et al.2011; Tian et al. 2014), particularly SNPs in some important motifs of the promoter part of a gene (Wang et al. 2017).However, this study focused on the CfC3hcoding region,and only a small part of the non-coding region was detected;thus, SNPs in 5′ UTR and 3′ UTR = regions will be sought in a further study.
Association analysis has been used to study the genetic architecture of important traits in forest. For example, Du et al. identif ied 202 signif icant SNPs in 63 candidate genes selected by transcriptome and QTL mapping that associated to plant growth (Du et al. 2016). In addition, dynamic association studies have been used to integrally identify the genetic basis of complex traits (Du et al. 2019). In future study, more association strategies will be undertaken to previously obtain important molecular markers to serve theC.fargesiibreeding.
Conclusion
In our study, we f irst cloned a putativeC3hhomologous gene inC. fargesiiand totally 163 SNPs were identif ied according to the alignment result from a mapping population including 88 naturalC. fargesiiindividuals. The LD decay distance short within theCfC3h(r2< 0.1 within 1800 bp). In additional, 8 SNPs and 10 haplotypes were identif ied signif icantly associated with 5 and 8 detected traits, respectively,using association a(h2) analysis. Our study implies allelic variations withinCfC3hmay inf luence wood properties ofC.fargesiiand the SNP markers identif ied in this study may be useful for marker-assisted selection, to improve wood traits inC. fargesiiin the future.
AcknowledgementsWe would like to appreciate Dr. Longxing Wang and Dr. Chenrui Gong for guiding the data analysis in this work.
杂志排行

Journal of Forestry Research的其它文章
- Genome-wide identif ication and cold stress-induced expression analysis of the CBF gene family in Liriodendron chinense
- Decay rate of Larix gmelinii coarse woody debris on burned patches in the Greater Khingan Mountains
- Characterizing conservative and protective needs of the aridland forests of Sudan
- Point-cloud segmentation of individual trees in complex natural forest scenes based on a trunk-growth method
- Accuracy of common stem volume formulae using terrestrial photogrammetric point clouds: a case study with savanna trees in Benin
- Appropriate search techniques to estimate Weibull function parameters in a Pinus spp. plantation