高效液相色谱法测定柴油多环芳烃含量的关键影响因素
2021-12-22宋春侠刘泽龙刘明星
宋春侠,刘泽龙,王 乾,刘明星
(1.中国石化 石油化工科学研究院,北京 100083;2.中国石化 炼化工程集团洛阳技术研发中心,河南 洛阳 471003)
柴油中多环芳烃不仅会影响柴油的燃烧性能,还会增加尾气中固体污染物和氮氧化物的含量,对环境和人体健康的危害较大[1-2]。为了更好地适应环境保护以及人们对清洁燃料的迫切需求,严格控制多环芳烃含量一直是车用柴油质量升级过程中的关键一环。目前,测定柴油中多环芳烃含量的标准方法主要有高效液相色谱法(SH/T 0806—2008)[3]、质谱法(SH/T 0606—2019)[4]、荧光指示剂法(GB/T 11132—2008)[5]和超临界流体色谱法(SH/T 0984—2019)[6]。其中,荧光指示剂法只能用于终馏点低于315 ℃,且无色的中间馏分;超临界流体色谱法则由于分析仪器较为昂贵、操作使用不方便等而应用较少。
作为目前柴油多环芳烃的主流分析方法,高效液相色谱法(SH/T 0806—2008)和质谱法(SH/T 0606—2019)各有优缺点。张大伟等[7]和张祎玮等[8]系统地考察了2种方法分析结果的差异性,发现模型化合物的选择会影响不同类型柴油芳烃含量测定结果的准确性。如加氢柴油中四氢萘类化合物含量明显高于烷基苯,此时如果采用烷基苯作为单环芳烃模型化合物,会导致单环芳烃的测定结果出现偏差,且偏差随着四氢萘类化合物含量的增加而变大。白正伟等[9]详细对比过EN 12916—2006[10]、SH/T 0806—2008以及SH/T 0606—2005方法对柴油芳烃含量的测定结果,发现,同样采用液相色谱定量原理的EN 12916—2006与SH/T 0806—2008方法的测定结果较为一致,而与SH/T 0606—2005方法的测定结果则存在一定的偏差。由于液相色谱和质谱法2种分析方法的定量原理不同,因此无法从准确性方面对二者的分析结果进行评价。
2016年,国Ⅵ车用柴油标准对多环芳烃的含量(质量分数)限制从国Ⅳ、国Ⅴ柴油的11%降至7%[11],其仲裁检测方法也从原来的SH/T 0606—2005更改为SH/T 0806—2008。然而,在SH/T 0806—2008仲裁方法推广应用过程中,仍然存在部分色谱柱单环芳烃与双环芳烃分界点无法识别、保留时间漂移、不同检测机构结果差异较大等现象,给柴油产品质量控制带来不确定性。
为了更好地适应清洁柴油的质量检测需求,提供更为准确的多环芳烃含量测定结果,保证仲裁方法的顺利推广,笔者针对高效液相色谱方法应用过程中的关键因素,分别考察了液相色谱固定相、系统平衡时间、基线漂移等关键分析条件对多环芳烃含量测定结果的影响,并针对高效液相色谱方法存在的问题提出对应的解决方案,从而确保标准方法的正确实施以及柴油多环芳烃含量测定结果的准确性,为清洁柴油产品质量控制提供科学依据。
1 实验部分
1.1 原料和试剂
环己烷,质量分数99.5%,北京化工厂产品;邻二甲苯,质量分数99%,天津市大茂化学试剂厂产品;1-甲基萘、二苯并噻吩,质量分数均为98%,百灵威公司产品;9-甲基蒽、六甲基苯、萘、菲,质量分数均为98%,伊诺凯公司产品;正庚烷,高效液相色谱纯,Fisher公司产品。
实验所用柴油样品包括:1#直馏柴油、2#直馏柴油、3#加氢精制柴油、4#加氢裂化柴油、5#加氢裂化柴油、6#催化裂化柴油,上述柴油样品均由中国石化石油化工科学研究院加氢研究室提供。
1.2 分析仪器及分析条件
高效液相色谱法(HPLC):采用美国Agilent公司生产的1260型高效液相色谱仪,配备示差折光检测器。色谱柱分别采用Agilent公司Zorbax氨基柱(以下简称色谱柱A)、Dikma公司的Inspire氨基柱(以下简称色谱柱B)、Waters公司的Spherisorb氨基柱(以下简称色谱柱C)、Agilent公司的Zorbax氰基柱(以下简称色谱柱D)、Dikma公司的Platisil氰基柱(以下简称色谱柱E)和Waters公司的Spherisorb氰基柱(以下简称色谱柱F)对柴油样品进行分析,上述6种色谱柱规格均为250 mm×4.6 mm×5 μm。按照SH/T 0806—2008标准方法进行芳烃含量测定。进样量10 μL,流动相体积流速为0.8~1.0 mL/min,柱温箱30 ℃,内置有六通阀。
气相色谱-质谱法(GC-MS):采用美国Agilent公司生产的6890-5977型气相色谱-四级杆质谱联用仪,按照SH/T 0606—2019标准方法要求对柴油进行饱和烃、芳烃分离,采用DB-1MS色谱柱对分离后的饱和烃和芳烃组分进行分析,检测条件为:进样量1 μL,体积分流比30∶1,EI电离源,离子源温度250 ℃,检测质荷比(m/z)范围为50~500。
2 结果与讨论
2.1 色谱柱的选择对分析结果的影响
作为色谱分离系统最核心的部件,一根填充良好、柱效较高的色谱柱对于实现复杂组分高效分离至关重要。SH/T 0806—2008标准方法对色谱柱的要求有3点:(1)填料为氨基键合(或者极性氨基/氰基键合)硅胶固定相;(2)实现系统性能验证溶液(SPS)中4种化合物的基线分离;(3)环己烷和邻二甲苯的分离度大于5。但在标准方法实际推广过程中,常常会遇到满足上述要求的色谱柱却无法对多环芳烃含量进行测定的现象,且不同实验室测定的芳烃含量结果偏差较大,难以对柴油品质进行判断。笔者选择市售的6种极性氨基/氰基键合的正相色谱柱,采用SPS溶液来考察其分离效果,性能评价结果如表1所示。
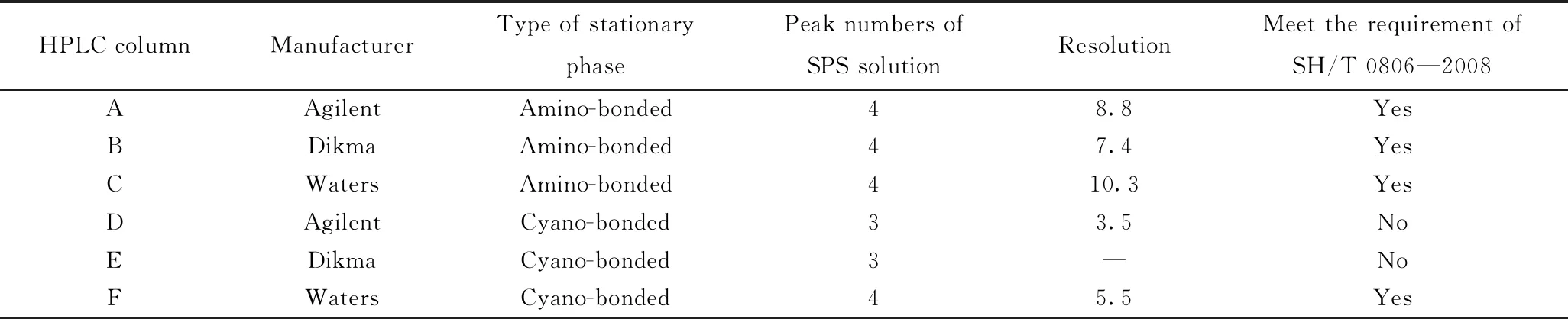
表1 6种极性氨基/氰基键合的正相色谱柱性能评价Table 1 Performance evaluation of six polar columns packed with amino/cyano-bonded stationary phase
从表1可以看出,色谱柱D和色谱柱E未能有效分离SPS溶液中的4种化合物,对环己烷和邻二甲苯的分离度均小于5,不符合SH/T 0806—2008标准方法对色谱柱分离度的要求,在后续研究中不再被选用。为进一步考察满足SH/T 0806—2008标准要求的色谱柱对实际柴油样品的分离效果,笔者选择6种不同类型的柴油,分别采用上述符合SH/T 0806—2008标准要求的4种色谱柱进行分离,典型的6#催化裂化柴油分离色谱图如图1所示。从图1可以看出,色谱柱A、B和F都可以按照极性的不同将柴油分成饱和烃、单环芳烃、双环芳烃和三环+芳烃4种组分,其测定的芳烃含量结果如表2所示。从表2中可知,对于大多数直馏柴油和加氢柴油样品而言,不同色谱柱芳烃含量测定结果差别不大,且均在方法再现性允差范围内。但对于芳烃含量较高的6#催化裂化柴油和3#加氢精制柴油而言,其多环芳烃含量差别显著高于其他类型的柴油样品(质量分数极差为4.3%)。
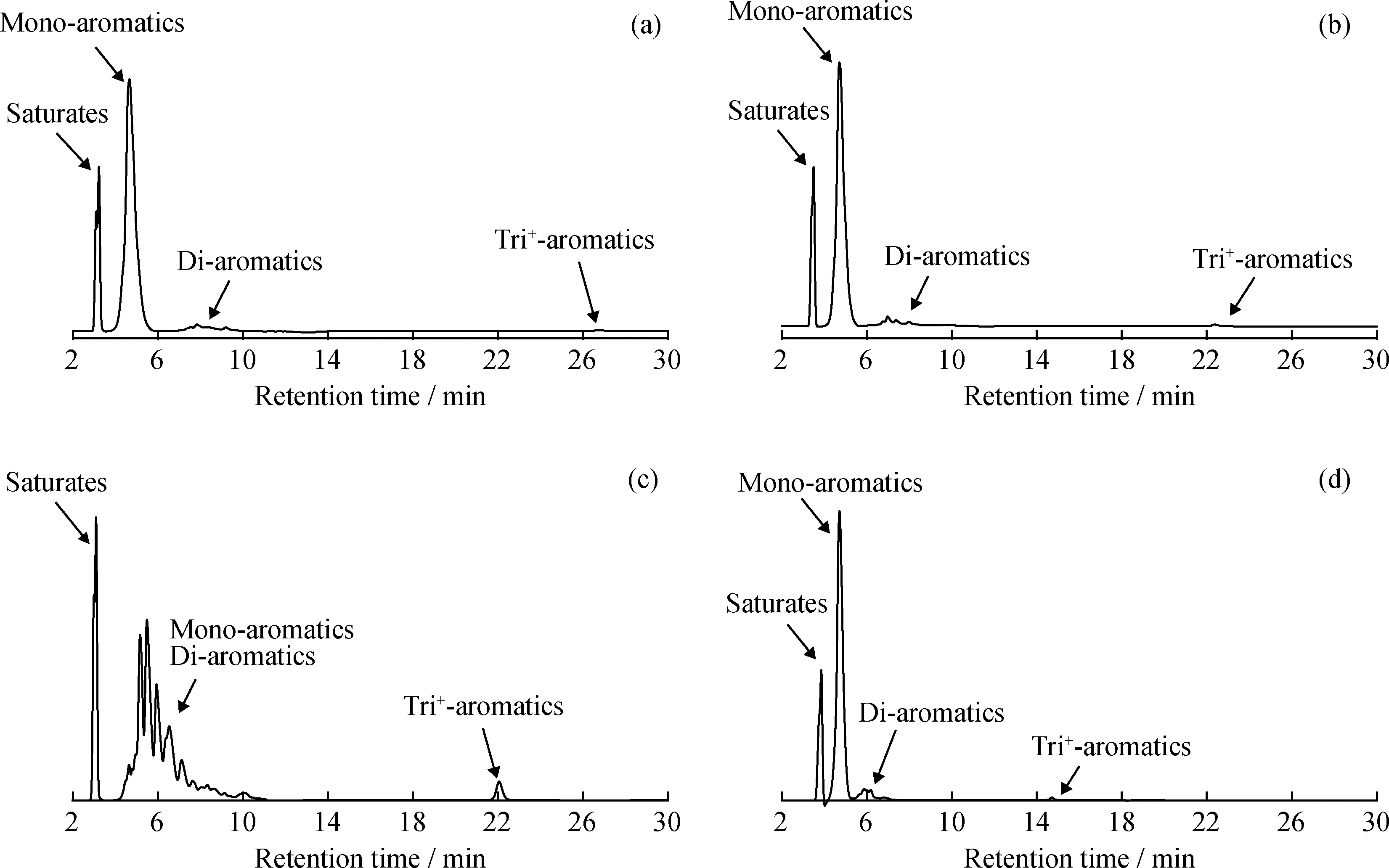
图1 符合SH/T 0806—2008标准要求的4种色谱柱对6#催化裂化柴油样品的分离色谱图Fig.1 Chromatograms of 6# FCC diesel fuel by four different HPLC columns which meet the resolution requirement of SH/T 0806—2008(a)Column A;(b)Column B;(c)Column C;(d)Column F Operating conditions:Mobile phase is 100% heptane;Flow rate is 1 mL/min
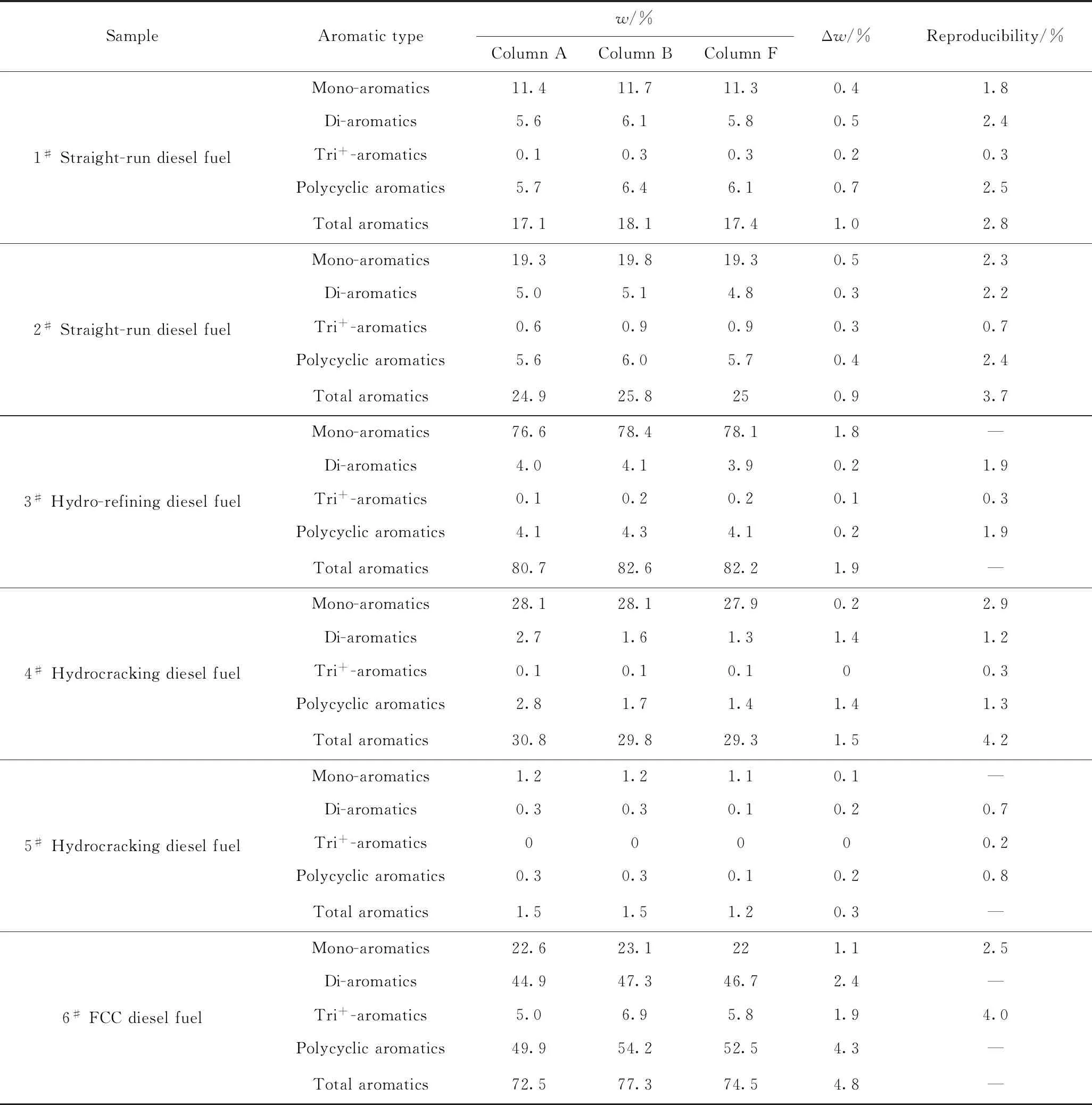
表2 3种类型色谱柱对6种柴油样品的芳烃测定结果对比Table 2 Determination results of aromatics in six different diesel fuels with using three different HPLC columns
张祎玮等[8]在考察柴油芳烃含量测定方法的适用性研究中也曾经发现,SH/T 0806—2008与SH/T 0606—2005标准方法测定的催化裂化柴油芳烃含量差别明显高于其他类型的柴油。从标准方法的适用范围来看,SH/T 0806—2008并未限制检测的柴油类型,但其精密度实验数据只适用于单环芳烃质量分数为4%~40%、双环芳烃质量分数为0~20%、多环芳烃质量分数为0~26%的柴油,而大多数催化裂化柴油中多环芳烃质量分数均大于30%。这说明如果采用高效液相色谱法测定催化裂化柴油或者其他芳烃含量较高的柴油样品,则无法根据精密度实验来判断芳烃检测结果的可靠性。另一方面,由于SH/T 0806—2008方法采用外标法进行芳烃含量的测定,其标准曲线的芳烃浓度范围与精密度实验样品的芳烃含量范围需保持一致,因此,如果需要对芳烃含量较高的柴油样品进行测定,则需要重新配制适用于高浓度芳烃含量的标准溶液,且要避免因芳烃含量过高引起的色谱柱过载情况,以确保芳烃含量测定结果的准确性。
由图1还可知,与色谱柱A、B和F相比,色谱柱C虽然可以将饱和烃和芳烃进行分离,但单环芳烃和双环芳烃组分却无明显唯一的峰谷,因此无法按照SH/T 0806—2008标准要求进行单环芳烃和双环芳烃分界点的划分,从而无法对柴油中的多环芳烃含量进行准确测定。为了排查色谱柱生产批次对分离效果的影响,笔者分别购买色谱柱C的不同批次产品进行分析实验,结果如图2所示,仍然无法对单环芳烃和双环芳烃分界点进行划分。大量实验结果表明,色谱柱C虽然分离度符合SH/T 0806—2008标准要求,但在柴油样品的分离过程中,确实存在由于单环芳烃和双环芳烃分界点不明显而无法对多环芳烃含量进行准确测定的问题。
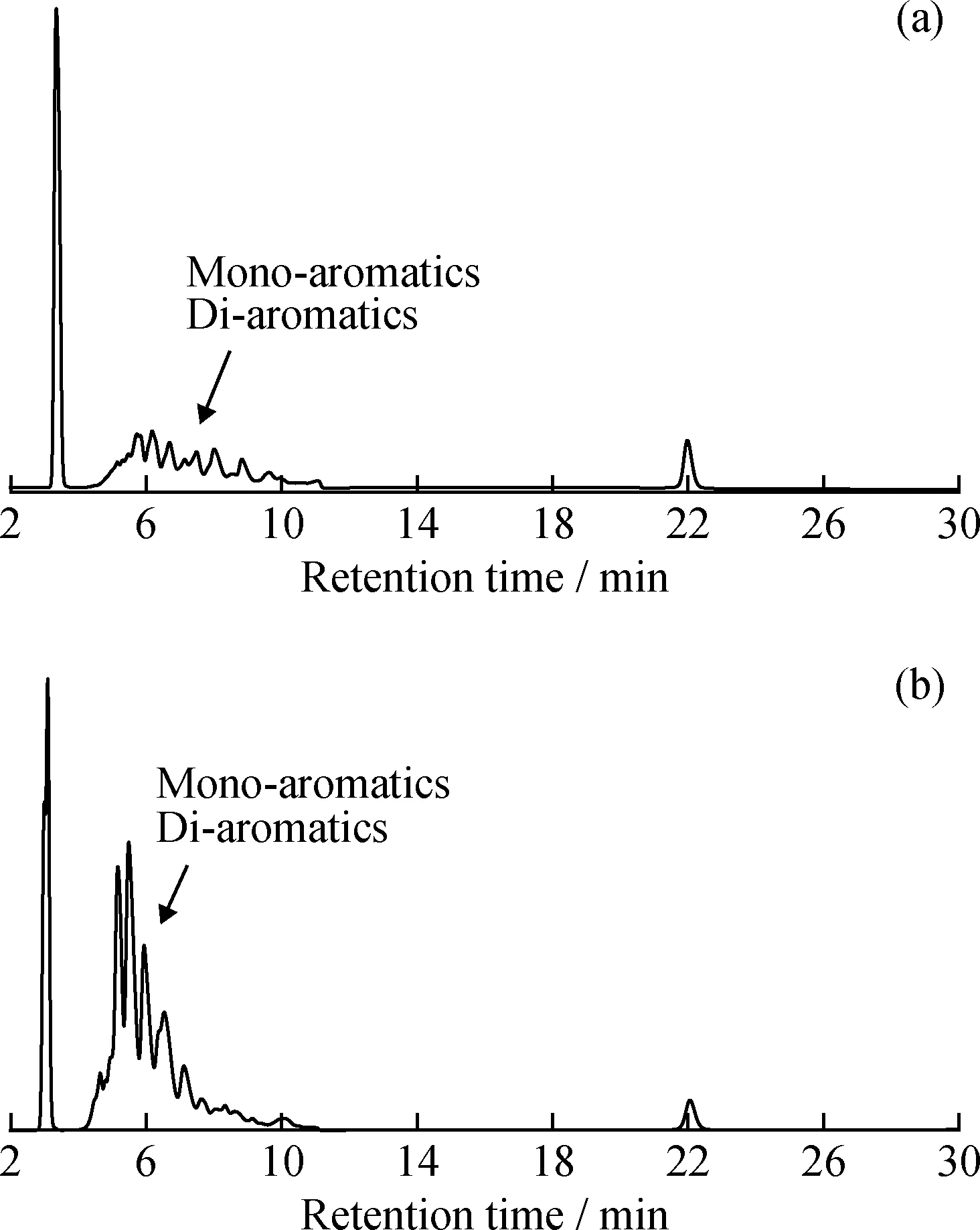
图2 不同生产日期的色谱柱C对6#催化裂化柴油样品的分离色谱图Fig.2 Chromatograms of 6# FCC diesel fuel by column C produced in the year of 2005 and 2017(a)2005;(b)2017 Operating conditions:Mobile phase is 100% heptane;Flow rate is 1 mL/min
SH/T 0806—2008是根据美国试验与材料协会标准ASTM D6591—2006进行起草的,值得注意的是,即使是在ASTM D6591—2019的最新修订版本中[12],色谱柱C(Waters公司Spherisorb系列的氨基柱)仍然为推荐的色谱柱系统,这主要是由于美国车用柴油产品标准中并没有对多环芳烃含量的限制[13],因此无需对单环芳烃和双环芳烃进行准确划分。但这一问题对于需要严格控制多环芳烃含量的中国车用柴油市场而言,则显得尤为重要,而这正是SH/T 0806—2008标准在应用过程中所需解决的核心问题之一。
为了进一步考察色谱柱C对不同类型芳烃的分离交叉情况,笔者将经过液相色谱分离的单环芳烃(F1)、双环芳烃(F2)和三环及以上芳烃(F3)组分进行收集,分别采用气相色谱-质谱法(GC-MS)对其组成进行分子识别,结果如表3所示。从表3可知,液相色谱分离得到的F1~F3组分中均存在与其他类型芳烃化合物不同程度的交叉现象。其中,进入F1组分的双环芳烃以萘为主,进入F2组分的单环芳烃以茚类(分子式为CnH2n-10)为主,而进入F3组分的双环芳烃以苊烯和芴类(分子式为CnH2n-16)为主,上述化合物的分子结构式如图3所示。从图3化合物的结构可以看出,虽然从芳烃类型划分来看,茚类和芴类/苊烯类分别属于单环芳烃和双环芳烃,但由于其本身的双环和三环结构,使其很容易在液相色谱分离过程中进入F2和F3组分,从而引起组分分离交叉重叠。
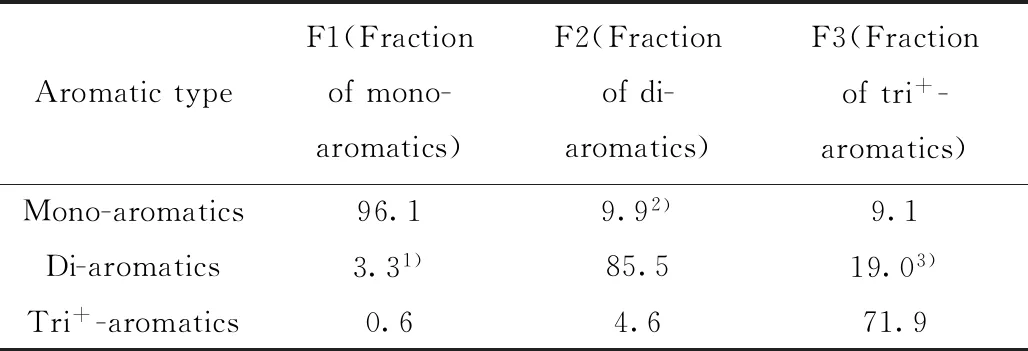
表3 液相色谱分离组分的芳烃类型分布Table 3 Distribution of aromatics for different fractions separated by HPLC w/%
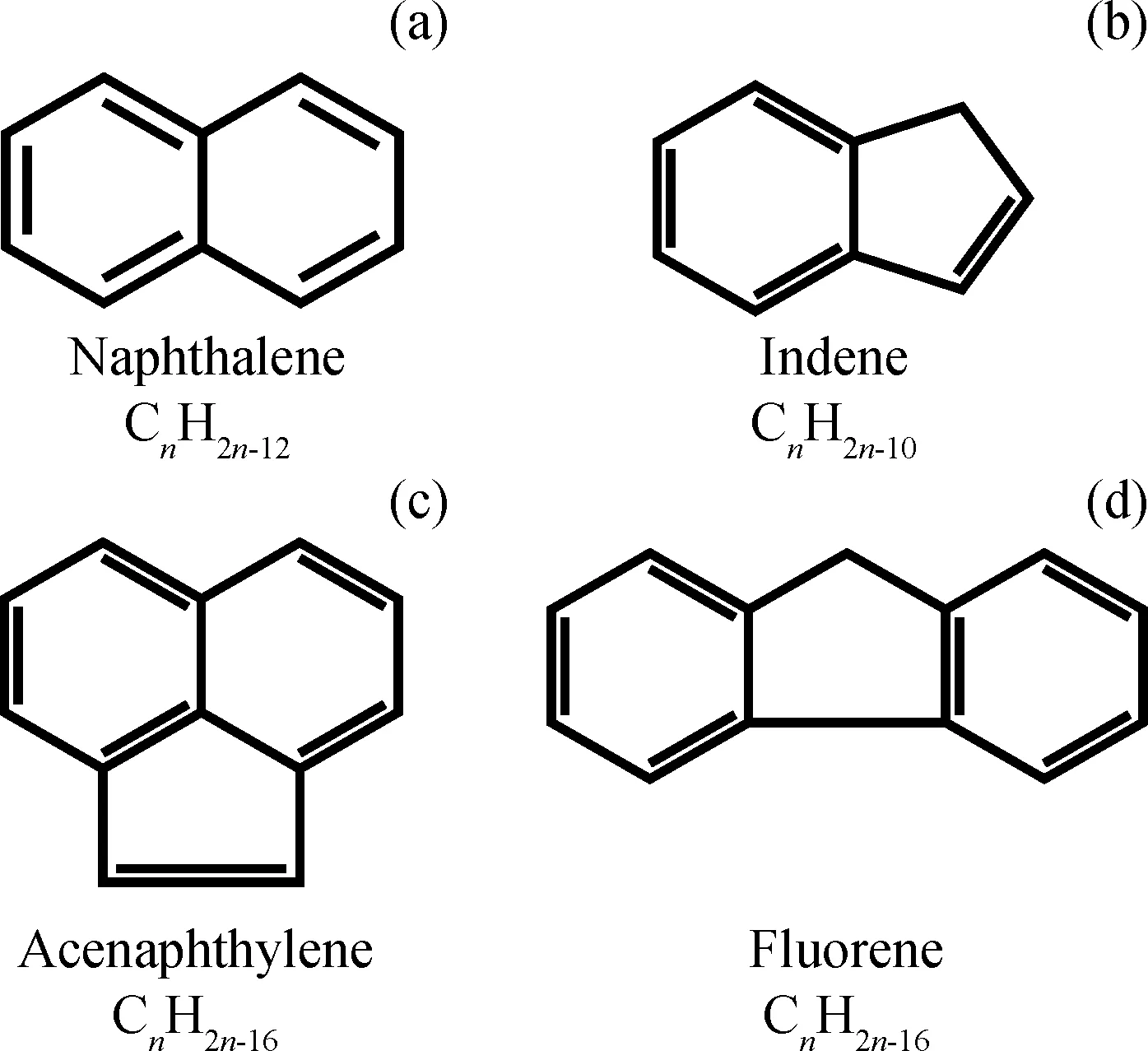
图3 典型芳烃化合物的结构式Fig.3 Structures of typical aromatic compounds
在影响芳烃分离结果的结构性因素中,除了环状结构的影响外,芳烃侧链的长短和个数也会引起芳烃极性的变化,从而带来芳烃组分的分离交叉重叠,这也是采用液相色谱法分离重馏分油更为困难的原因所在[14]。笔者以六甲基苯和萘为模型化合物,考察了这2种化合物在4种类型色谱柱上的分离效果,结果如图4所示。由图4可知,在色谱柱C中双环芳烃萘先出峰,而单环芳烃六甲基苯后出峰。这与SH/T 0806—2008中要求单环芳烃先出峰、双环芳烃后出峰的顺序相反,而其他3种色谱柱均不存在这一问题。
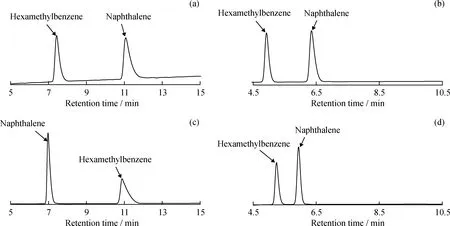
图4 六甲基苯和萘在4种色谱柱上的分离色谱图Fig.4 Chromatograms of hexamethylbenzene and naphthalene by four different HPLC columns(a)Column A;(b)Column B;(c)Column C;(d)Column F Operating conditions:Mobile phase is 100% heptane;Flow rate is 1 mL/min
综上所述,以色谱柱C(Waters氨基柱)为代表的色谱柱,虽然符合SH/T 0806—2008标准对色谱柱分离度的要求,但在实际应用过程中,由于其对六甲基苯和萘的洗脱顺序与标准方法要求的单环芳烃先洗脱、双环芳烃后洗脱的顺序不符,且其分辨率较高(环己烷和邻二甲苯的分离度大于10,表1)导致芳烃色谱峰明显多于其他类型色谱柱(如图1(c)所示),从而带来芳烃分界点不明确的问题,影响多环芳烃的准确测定。这说明SH/T 0806—2008标准中对于色谱柱系统的要求并不完善,需要对其进行重新修订。
实际上,从键合相液相色谱分离模式的特点来看,由于固定相是通过化学反应将有机官能团键合到硅胶颗粒表面的,这就意味着不同生产厂家,甚至同一厂家不同批次生产的固定相,其表面官能团覆盖情况都会有所差别[15],而这势必会对色谱柱分离结果造成影响。为了避免这一问题,就需要对色谱柱系统进行较为严格的使用条件限制,使其分离效果在很小的范围内波动,从而得到更为准确、一致的定量分析结果。
值得注意的是,同样采用高效液相色谱法分离,外标法定量的GB/T 25963—2010标准[16]也采用极性氨基/氰基键合的色谱柱对中间馏分油芳烃含量进行测定。与SH/T 0806—2008标准所不同的是,该标准对SPS溶液的洗脱顺序有要求(按照环己烷、十二烷基苯、邻二甲苯、六甲基苯、萘、二苯并噻吩和9-甲基蒽的先后顺序进行洗脱),且规定环己烷和邻二甲苯的分离度要在5.7~10之间,按照这2点限制条件,都可以将Waters氨基柱(色谱柱C)排除在外。因此,在未来SH/T 0806—2008标准方法修订过程中,如果能够与GB/T 25963—2010标准方法对色谱柱的要求保持一致,则可以有效排除特殊色谱柱C对芳烃测定结果的影响。
2.2 反冲洗时间对分析结果的影响
为节省色谱分析时间,SH/T 0806—2008方法采用反冲洗方案将保留能力较强的三环及以上芳烃反冲洗出来,其反冲洗时间根据SPS溶液中的二苯并噻吩和9-甲基蒽的保留时间来确定。由于正相色谱柱所需平衡时间较长,因此,二苯并噻吩和9-甲基蒽的保留时间会随着色谱柱平衡时间的延长而不断变化,从而引起反冲洗时间的不断波动。以色谱柱F为代表,笔者采用SPS溶液考察了其获得稳定反冲洗时间的平衡体积,结果如图5所示。由图5可知,色谱柱F平衡所需溶剂大约为40~60倍的柱体积(约为120 ~180 mL)才能获得满意的分离度以及可重复的保留时间。其他色谱柱达到稳定反冲洗时间所需的平衡体积与色谱柱F较为接近,且色谱柱放置不用的时间越长,所需的平衡体积越大,新色谱柱初次平衡有时甚至需要1~2 d。
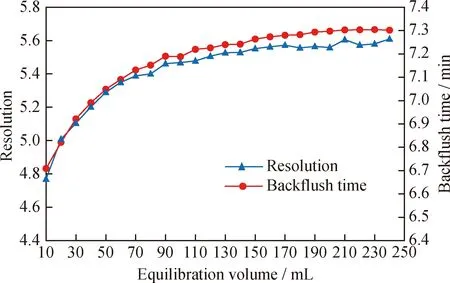
图5 色谱柱F的分离度及反冲洗时间随平衡体积变化的波动情况Fig.5 Changes of resolution and backflush time of column F with different equilibration volumesOperating conditions:Mobile phase is 100% heptane;Flow rate is 1 mL/min
对于正相键合相色谱而言,流动相中的水含量会极大地影响色谱柱的分离性能[14],从而带来多环芳烃含量测定结果的不确定性。由于环境湿度变化的不可控性,即使通过严格的除水系统得到超干溶剂,也很难保证放置一段时间后流动相的水含量保持不变。因此,在采用液相色谱法对柴油样品进行分离前,需要通过SPS溶液监测色谱柱保留时间的重现性,确保色谱柱被流动相充分平衡,从而获得准确的反冲洗时间。
为了考察不准确的反冲洗时间对多环芳烃含量测定的影响,笔者首先采用模型化合物进行实验验证,结果如图6所示。从图6可以明显看出,延后反冲洗时间会造成化合物菲被分隔为2部分,从而影响多环芳烃测定结果的准确性,同样的结果也在柴油样品应用中被验证,结果如表4所示。由表4可知:如果提前1 min切换反冲洗阀,会使得部分双环芳烃被冲洗到三环及以上芳烃组分中,从而造成双环芳烃含量偏低(与正常切阀时间得到的芳烃含量相比,下同),三环及以上芳烃含量偏高。如果延后1 min切换反冲洗阀,会使得部分三环及以上芳烃被冲洗到双环芳烃组分中,从而造成双环芳烃含量偏高,三环及以上芳烃含量偏低的现象。且色谱柱需要预平衡的时间越长,不正确的反冲洗时间对芳烃含量测定结果的影响就越大。这说明采用SH/T 0806—2008标准方法进行实际样品测定前,必须采用SPS溶液对色谱柱平衡状态进行准确的评估,一般推荐采用正庚烷平衡2 h以上再进行实际柴油样品的测定。
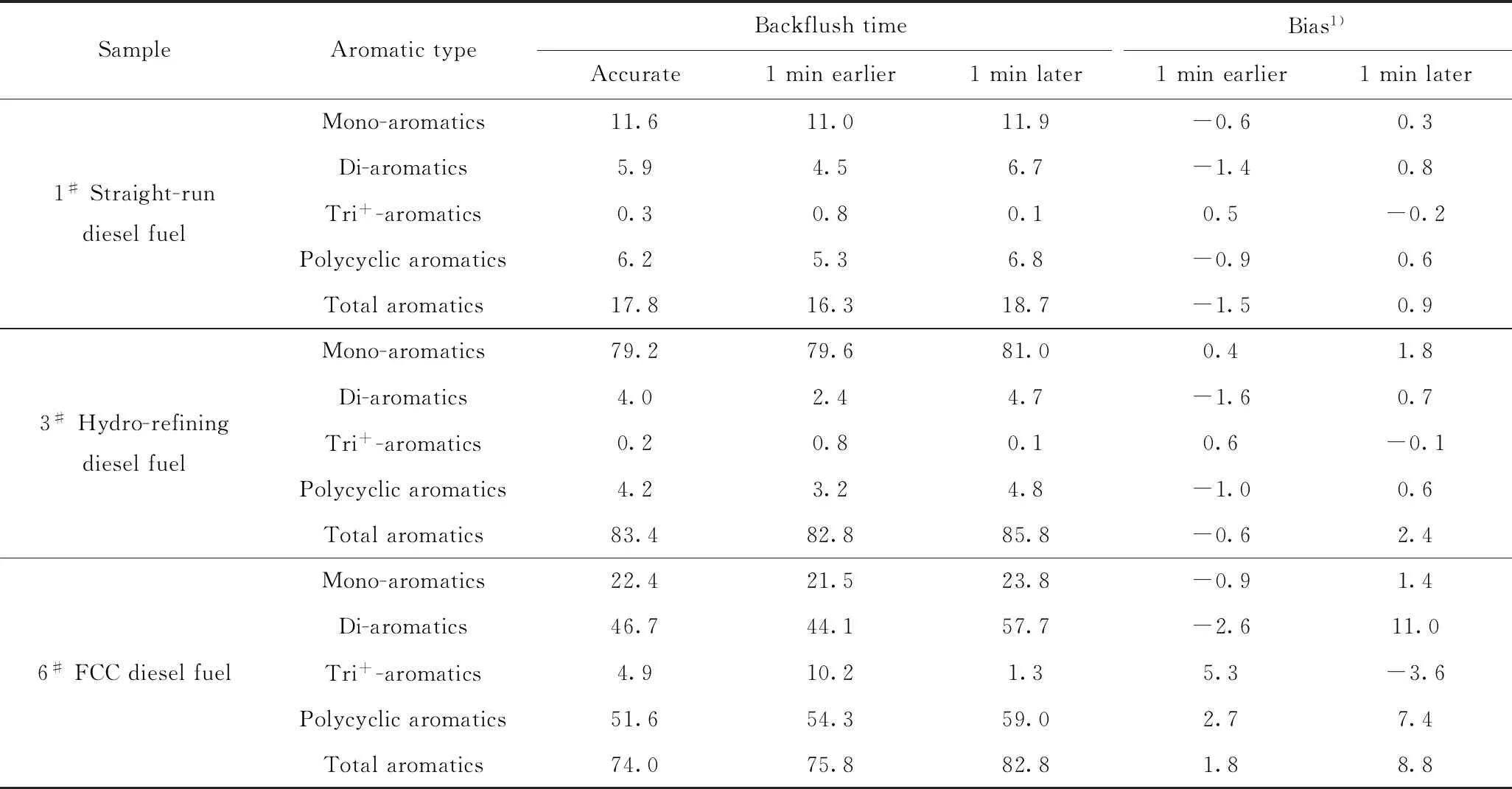
表4 反冲洗时间对色谱柱F中芳烃含量测定结果的影响Table 4 Effects of different backflush time on aromatics determination results by column F w/%
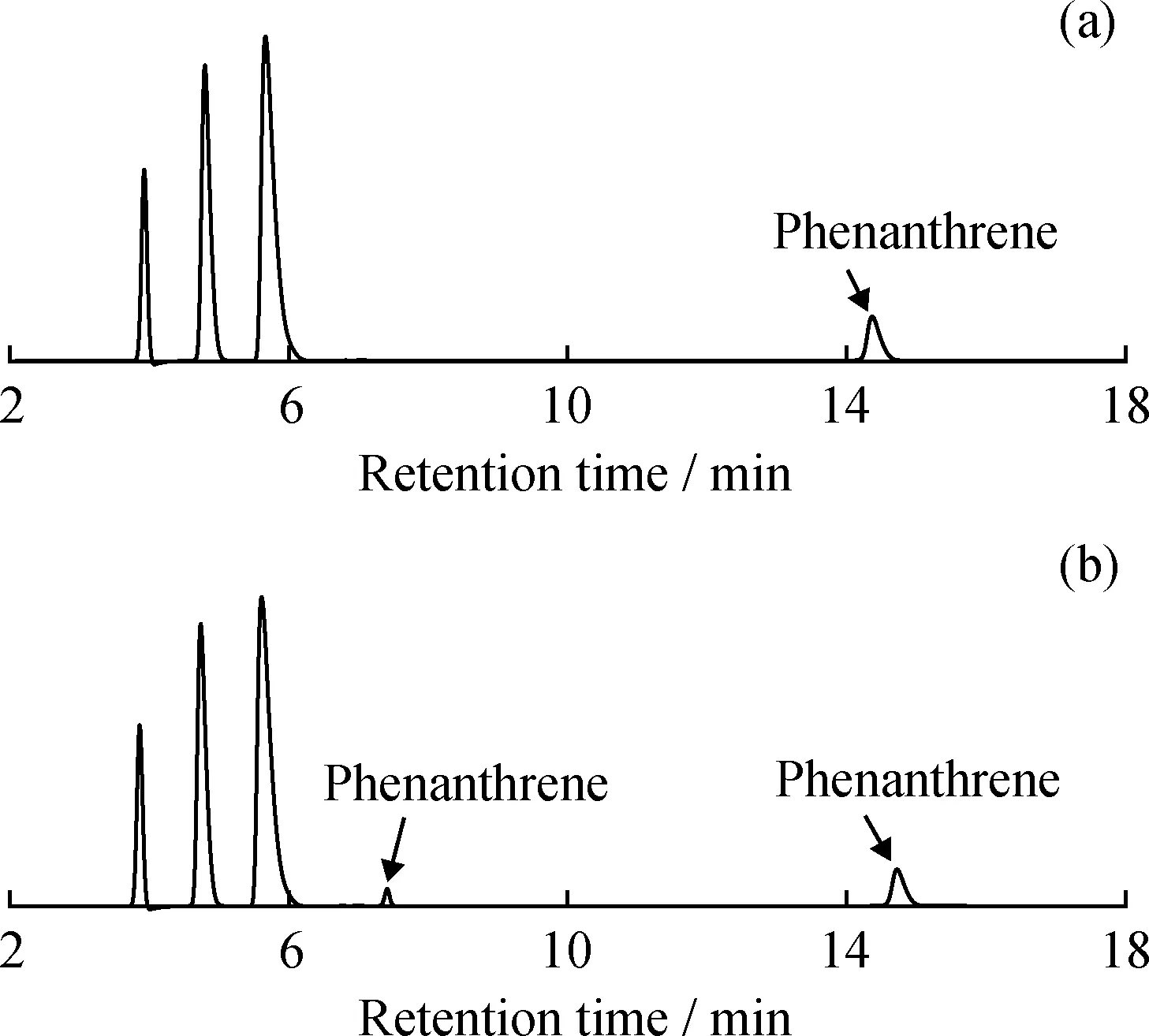
图6 反冲洗时间对色谱柱F中模型化合物分离的影响Fig.6 Effects of different backflush time on model compounds separation by column F(a)Accurate backflush time;(b)Delayed backflush timeOperating conditions:Mobile phase is 100% heptane;Flow rate is 1 mL/min
2.3 基线漂移对芳烃含量分析结果的影响
作为液相色谱的通用型检测器,示差折光检测器对温度、压力的变化都极为敏感,一旦引起基线漂移,必然会引起色谱峰面积的波动,从而影响芳烃含量的测定结果。笔者发现一些多环芳烃含量较高的样品,在反冲洗阀切换处会发生较为明显的基线漂移现象。虽然符合SH/T 0806—2008标准中所规定的“不超过环己烷峰高的0.5%”,但由于该分界点的选择直接影响了芳烃色谱峰的基线,在某些情况下会带来非常大的影响。
以6#催化裂化柴油在色谱柱F上的分离色谱图(如图7所示)为例,考察基线漂移对芳烃含量结果的影响。由图7可知:根据SPS溶液分离时计算得到的反冲洗时间为7.3 min(E点),如果从非芳烃开始处(A点)到E点(7.3 min)作基线,则会由于峰谷D点(7.2 min)的存在而导致整体基线偏高(蓝线);如果从A点到峰谷F点(7.5 min)作基线,虽然可使整体基线保持一致,但又不符合SH/T 0806—2008方法关于“到反冲洗阀将要开始动作处作基线”的要求。针对这一问题,笔者建议应该从A点到峰谷D点(7.2 min)作基线,这样既符合标准对基线选取的要求,又不会因为引入负峰而导致芳烃峰面积减小的情况。分别以D点、E点和F点作为双环芳烃的终点,计算样品的芳烃含量,结果如表5所示。从表5可知,对于芳烃含量较高的催化裂化柴油样品而言,双环芳烃终点的选择可能会引起多环芳烃含量近10%的偏差。因此,在测定此类样品时,需要格外注意基线漂移对芳烃含量结果的影响。
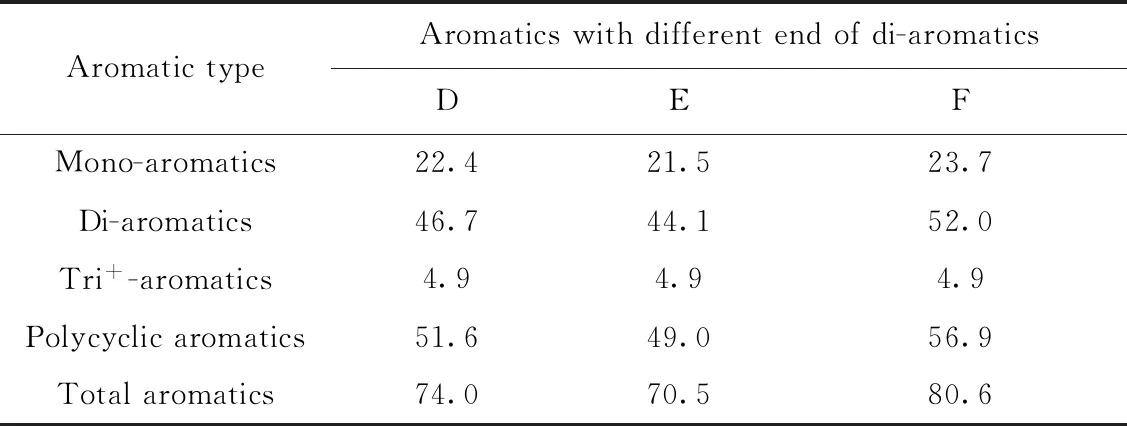
表5 基线漂移对6#催化裂化柴油芳烃含量测定的影响Table 5 Effects of baseline drift on aromatics determination results in 6# FCC diesel fuel w/%
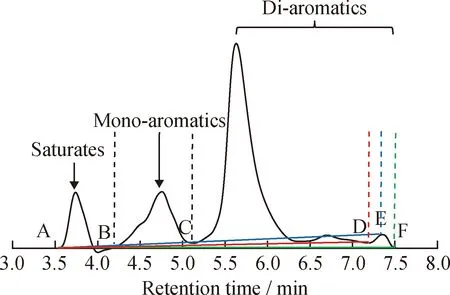
A—The beginning of saturates;B—The beginning of mono-aromatics;C—The beginning of di-aromatics;D—The valley before the backflush point;E—Backflush point;F—The valley after the backflush point图7 基线漂移对6#催化裂化柴油芳烃含量测定的影响Fig.7 Effects of baseline drift on aromatics determination results in 6# FCC diesel fuelOperating conditions:Column F;Mobile phase is 100% heptane;Flow rate is 1 mL/min
对于大多数加氢柴油、直馏柴油以及车用柴油来说,由于其多环芳烃含量较低,在反冲洗阀切换处并不会引起基线较明显的波动,因此基线的划分并不会对其芳烃含量测定结果造成显著影响。这也从另一方面证实了,与催化裂化柴油相比,液相色谱法可能更适用于测定成品柴油、加氢柴油以及直馏柴油的多环芳烃含量。
2.4 SH/T 0806—2008与SH/T 0606—2019测定芳烃含量结果的对比
在GB 19147—2016标准颁布之前,SH/T 0606—2005标准(质谱法)一直作为柴油多环芳烃含量测定的仲裁方法被采用。在新旧产品标准过渡时期,2种标准都被广泛用于车用柴油中多环芳烃含量的测定,但2者的测定结果常常存在较大的偏差,这往往会给车用柴油的质量控制带来较多的问题。
因此,笔者选择不同类型的柴油样品,分别采用SH/T 0606—2019与SH/T 0806—2008标准方法进行测定,结果如表6所示。从表6中可知,SH/T 0606—2019与SH/T 0806—2008标准方法得到的芳烃含量差别较大。与SH/T 0606—2019标准方法相比,SH/T 0806—2008标准方法得到的单环芳烃含量普遍偏高,而多环芳烃含量偏低,且部分样品总芳烃质量分数偏差超过10%,这一规律与张大伟等[7-8]的研究结果相一致。这说明对于部分柴油样品而言,采用SH/T 0606—2019标准还是SH/T 0806—2008标准方法对其芳烃含量进行测定,将会极大影响着对柴油品质的判定。
实际上,由于柴油样品组成复杂,化合物侧链个数以及环结构的不同会给其样品的分离带来极大的不确定性。从2.1小节不同组分的分离交叉也可以看出,无论是基于吸附色谱原理的SH/T 0606—2019标准方法还是基于正相键合相色谱的SH/T 0806—2008方法,都难以避免相邻组分的分离交叉重叠。与单纯依靠色谱峰面积定量的SH/T 0806—2008方法所不同的是,SH/T 0606—2019方法后续可以通过质谱特征碎片对饱和烃和芳烃组分中的分离交叉部分进行校正,因此,其总芳烃的定量结果相对更为准确。
大量的研究结果表明[7-8],对于不同类型的柴油而言,由于其芳烃组成差别较大,因此选择一种模型化合物来代替一类芳烃,可能会使芳烃测定结果出现偏差。如表6中3#加氢柴油是以催化裂化柴油为原料通过加氢精制而获得,其单环芳烃主要以四氢萘类为主,此时如采用烷基苯作为单环芳烃的模型化合物,会使其单环芳烃质量分数的测定值与SH/T 0606—2019标准方法相差14%。因此,对于催化裂化柴油加氢后得到的柴油样品,并不适合采用烷基苯进行单环芳烃工作曲线的绘制。
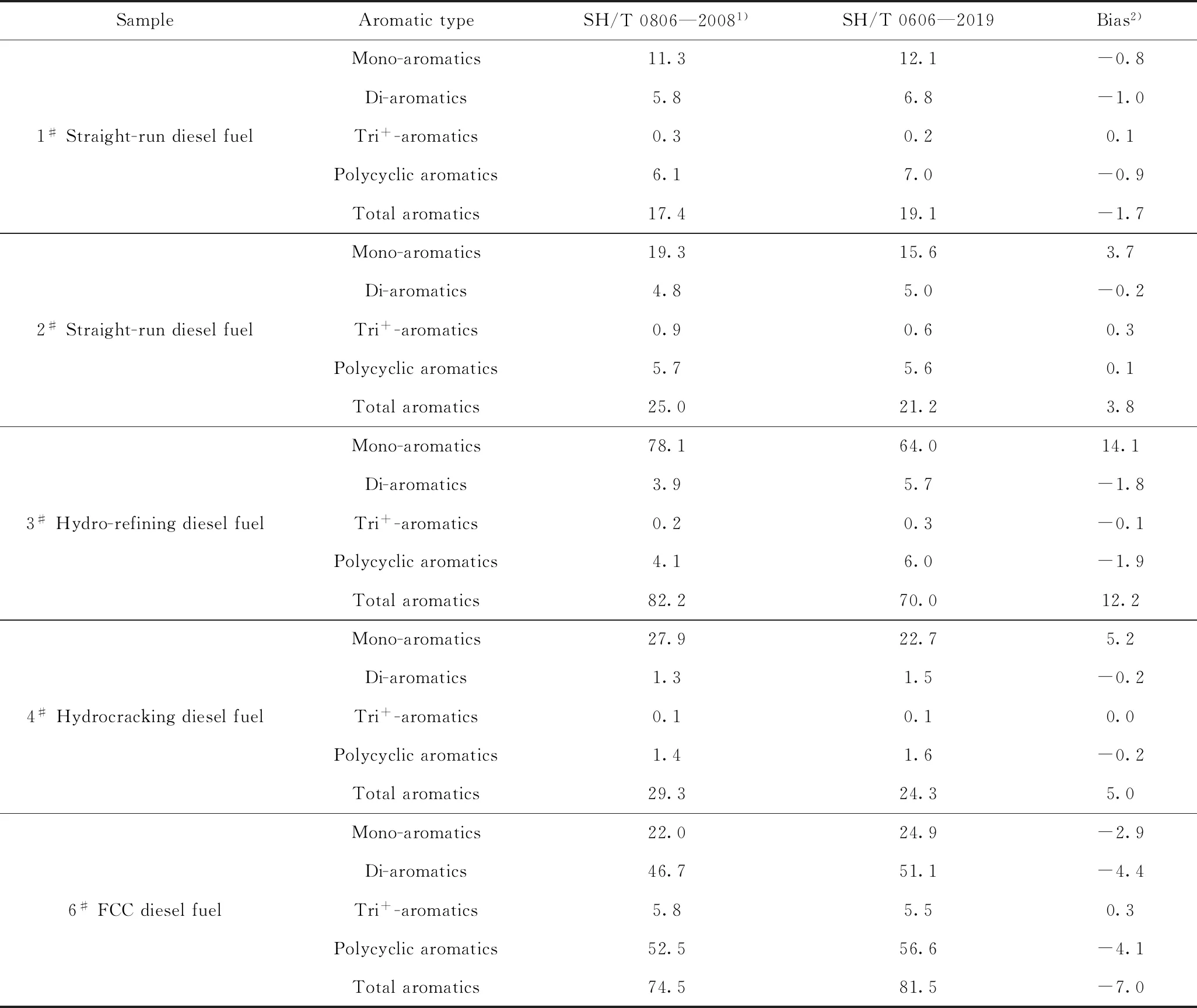
表6 SH/T 0806—2008与SH/T 0606—2019方法的对比Table 6 Comparison of determination results between SH/T 0806-2008 and SH/T 0606—2019 methods w/%
通过前面的分析可以看出,SH/T 0806—2008方法并不适用于芳烃含量较高的催化裂化柴油以及其经加氢处理后的柴油,且SH/T 0806—2008方法的精密度范围并未包括此类型的柴油样品。因此,对于芳烃含量较高的柴油,推荐采用质谱法对其进行测定,以避免因方法选择不当而对产品质量判定产生影响。
3 结 论
(1)影响高效液相色谱法测定柴油多环芳烃结果准确性的关键因素有:固定相的类型,反冲洗时间的确认、色谱基线以及模型化合物的选择。
(2)为得到准确、可靠的芳烃含量测定结果,需要注意如下4点:第一,部分氨基键合的液相色谱柱,在实际柴油样品分析中存在芳烃分界点不明确的问题,影响多环芳烃的准确测定,不建议采用;第二,选择高效液相色谱纯的正庚烷溶剂做流动相,且在使用前进行除水脱气处理;第三,样品分析前需采用SPS溶液测试系统状态,待反冲洗时间波动在0.2 min内方可进行样品测试;第四,保持检测器以及色谱柱分析系统的温度稳定,且参考池充满最新的流动相,确保基线漂移不超过环己烷峰高的0.5%;
(3)对于芳烃含量较高的催化裂化柴油以及部分加氢精制柴油,推荐采用SH/T 0606—2019标准方法进行多环芳烃含量的测定。
