自组装超分子前驱体制备管状氮化碳及模拟太阳光光催化降解水中双氯芬酸
2021-12-21刘晓娜黄韬博钱天伟
刘晓娜, 黄韬博, 陈 龙, 李 博, 钱天伟, 李 璠*, 刘 文
1.太原理工大学环境科学与工程学院, 山西 太原 030024 2.北京大学环境科学与工程学院, 水沙教育部重点实验室, 北京 100871 3.北京大学, 国家环境保护河流全物质通量重点实验室, 北京 100871
随着社会经济的发展,大量新有机污染物被排入水中,如药物及个人护理品、内分泌干扰物、多氟/全氟烷基类化合物等. 新有机污染物具备环境持久性、高生物活性、高环境毒性等特征,且无法被污水处理厂常规技术去除,已成为热点问题[1-4]. 活性药物中,双氯芬酸(DCF)作为最流行的非甾体抗炎药之一,在世界范围内广泛使用[1-3]. 人体代谢后,DCF进入城镇污水处理系统,因DCF生物降解率低,无法高效去除,可在污水处理厂出水口和地表水中被检出[3-5]. 虽然自然环境中DCF检出浓度处于较低水平(μg/L或ng/L)[6],但DCF较高的辛醇-水分配系数(lgKOW≈4.5)[7]导致其可通过食物链富集在生物体内,增加DCF生物暴露风险. DCF引发的人类健康和生态系统风险引起了人们的关注[8-9],开发高效去除DCF的新技术至关重要. 常用水中DCF去除技术包括吸附法和高级氧化法. 使用活性炭[10]、碳纳米管[11]、碳纤维[12]、金属有机框架[13-14]和离子交换树脂[15]等去除DCF存在以下明显弊端:① DCF不能降解脱毒,存在二次污染风险;②再生成本高,需使用昂贵的有毒溶剂[16-17].
高级氧化工艺(AOPs)已被证实是有机污染物去除的最有效技术之一[18-21]. 因太阳光驱动的光催化技术利用清洁的太阳能,并具备高效的去除能力,成为最具前途的AOPs技术之一[22-23]. 目前,大量光催化剂(TiO2基[23]、Bi基[24]和Ag基[25])被应用于DCF光催化降解. 石墨相氮化碳(g-C3N4,GCN)作为一种二维非金属半导体,因具有突出的可见光吸收能力、高热稳定性、高化学稳定性以及特殊的电子结构,受到越来越多关注[26-28]. GCN优异的光电性质使其适用于多种场景,如光催化析氢反应(HER)、氧还原反应(ORR)和析氧反应(OER)等. 但是,直接热聚合法制备的体相型g-C3N4存在明显缺点,如表面积低和活性位少[1,28],极大限制了其应用. 此外,GCN的可见光量子效率也需进一步提升. 构建GCN的分层微、纳米结构和异质金属元素掺杂是提升GCN光催化活性并解决上述问题的有效方法[29]. 异质金属元素可能会造成有毒金属析出,引发环境风险,故构建GCN分层微、纳米结构成为GCN改良的首选绿色手段,如通过硬模板法已成功构建多孔、球形和管状等结构的GCN[29].
由于氢键具有较强的方向性和饱和性,以三聚氰酸-三聚氰胺配合物为原料,通过分子间氢键自组装形成超分子前驱体,可制备具特殊形貌与结构的GCN[29-31]. 使用多种原料(如尿素、二氰二胺、三聚氰胺、三聚氰酸等)在有机溶剂中组装超分子前驱体,并通过控制合成条件制备空心结构[32]、微球状[33]和片状的GCN[34]. Ong等[35]研究发现,水溶剂更利于分子自组装,形成大尺寸稳定规则的超分子前驱体,水热条件下三聚氰胺可转化为三聚氰酸,并与三聚氰胺原位自组装形成超分子前驱体.
该研究采用水热法将三聚氰胺部分转化为三聚氰酸,通过原位自组装,制备超分子前驱体,再以热聚合方法最终合成比表面积大、活性位点丰富的管状g-C3N4(TCN). 此外,以活性药物DCF作为目标污染物,探究了TCN对DCF光催化降解的性能及机理,以期为调控合成具有高太阳光响应的氮化碳材料以及高效去除水中新有机污染物的应用提供设计思路与理论支持.
1 材料与方法
1.1 材料与试剂
双氯芬酸钠购自Sigma-Aldrich (St. Louis, MO, 美国),氢氧化钠、高氯酸、三聚氰胺、亚磷酸、碘化钾、异丙醇(IPA)、对苯醌(BQ)等试剂购于国药控股有限公司. 上述试剂均为试验分析级别,使用前未做进一步纯化.
1.2 催化剂合成方法
GCN合成方法采用一步热聚合法合成[26-27,31]:将3 g三聚氰胺装入坩埚(10 mL)形成半密封状态后置于马弗炉中,以5 ℃/min加热至550 ℃,维持3 h.
自组装超分子前驱体制备:将1 g三聚氰胺与0.6 g亚磷酸混合,加入50 mL去离子水,在60 ℃下快速磁力搅拌0.5 h. 待亚磷酸完全溶解且三聚氰胺在溶液中分散均匀后,将混合液转移至含特氟龙内胆的不锈钢反应釜(总体积 80 mL)中,180 ℃水热10 h,用去离子水清洗自组装超分子前驱体3次,乙醇清洗1次,真空干燥箱中60 ℃烘干后用于后续TCN合成.
TCN制备:称取3 g自组装超分子前驱体装入坩埚(10 mL)后,放入管式炉,在氩气的保护下(氩气流速为10 mL/min),以2 ℃/min加热至550 ℃,保持3 h.
1.3 材料表征
使用FEI Nova Nano SEM 450(美国)场发射扫描电镜(FESEM)表征催化剂的表面形貌,工作电压为5 kV. 使用IRTracer-100光谱仪(岛津,日本)测量样品的傅里叶变换红外光谱(FT-IR),以KBr为背景,制样中KBr与样品质量比为 200∶1,扫描范围为450~3 800 cm-1. 使用Dmax-2400 Rigaku型(Rigaku,日本)X射线衍射仪(XRD)对材料晶型结构进行表征,在100 kV、40 mA、Cu Kα辐射(λ=1.542 Å)条件下,选取5°/min扫描步长测样. 使用ASAP-2010比表面积分析仪(Micromeritics, 美国)测量材料的Brunauer-Emmett-Teller (BET)比表面积,同时基于Barret-Joyner-Halender (BJH)法得到孔径分布. 使用岛津UV-3200分光光度计(日本)测量材料漫反射紫外-可见吸收光谱(UV-vis DRS),并根据Kubelka-Munk变换评估能带间隙(Eg). 使用配备泊菲莱光源系统(中国)的电化学工作站(Metrohm Autolab PGSTAT204)分析材料光电性能,测量使用标准三电极体系,Pt为对电极,Ag/AgCl为参比电极. 工作电极上样品制备方法:先将2.5 mg催化剂和50 μL的Nafion®溶液均匀分散在0.45 mL乙醇溶液中,再将材料(20 μL)涂在1 cm×1 cm的氧化氟锡(FTO)玻璃电极上,待其自然干燥. 电解液为0.5 mol/L Na2SO4. 电化学阻抗谱测试的试验在开路电位振幅为5 mV下进行,频率范围为10-2~104Hz. 莫特肖特基也在该体系下测量,用于计算催化剂的平带电位,测定的频率为 1 000 Hz. 在0.5 mol/L的氮气饱和Na2SO4溶液中,以20 mV/s的扫描速率进行线性扫描伏安测试.
1.4 双氯芬酸降解试验
光催化试验在300 W氙气弧光灯光源(PLS-SXE300D,泊菲莱,中国)下进行,采用模拟太阳光AM 1.5模式,反应器悬浮液表面光强为(120±5)mW/cm2. 循环水浴控制反应温度为(23.0±0.5)℃. 试验中将100 mg光催化剂分散在100 mL DCF溶液(浓度为10 mg/L)中,用0.1 mol/L NaOH或高氯酸溶液将悬浊液调至设定pH. 在暗室条件下搅拌30 min,使DCF在光催化剂上达到吸附平衡. 在持续光照一定时长后采样,收集1 mL悬浮液与1 mL甲醇进行混合,并用0.22 μm尼龙膜分离材料,采用高效液相色谱(HPLC,Agilent 1260,美国)检测DCF浓度. HPLC配备Agilent XDB-C18柱(2.1 mm×100 mm, 3.5 μm),测样温度为30 ℃,紫外检测器波长为276 nm,流动相体积分数为70%乙腈和30%水(含0.2%乙酸),流动相流速为1 mL/min. 考察溶液pH对TCN光催化降解DCF的影响,调pH至5、7和9,反应时间为60 min.
1.5 活性物种检测与鉴定
通过淬灭剂试验间接研究不同活性物种在光催化反应中的贡献,分别以异丙醇〔IPA,淬灭动力学k=1.9×109(M·s)-1〕、碘化钾〔KI,淬灭动力学k≥1.1×1010(M·s)-1〕和对苯醌〔BQ,淬灭动力学k=3.7×106(M·s)-1〕淬灭反应体系中的羟基自由基(·OH)、光生空穴(h+)和超氧根自由基(·O2-)[16]. 3种淬灭剂投加剂量均为10 mmol/L. 使用电子自旋共振检测·OH和·O2-的生成,在布鲁克EMXplus(德国)仪器ER-4119-HS高灵敏度垂直模腔上获取X-band 频率为9 GHz的电子自旋共振谱图. 电子自旋共振运行条件:中心场为 3 510 G;扫描宽度为100 G;调制频率为100 kHz;微波功率为20 mW. 进行5~10次扫描后获得可靠的信噪比. 试验使用的自旋捕集剂为5,5-二甲基-1-吡咯啉-N-氧化物(DMPO):将100 μL DMPO加入100 μL催化剂悬浮液(1 g/L)后,模拟太阳光照射60 min制备样品,将其放入圆柱形石英反应单元中以测量电子自旋共振谱图.
1.6 催化剂重复利用试验
通过5个连续的循环光催化试验探究TCN的重复利用性能. 循环利用试验中,光催化反应时间为1 h.
2 结果与讨论
2.1 材料表征与分析
GCN与TCN的FT-IR谱图〔见图1(a)〕显示: TCN与GCN出峰无显著差异,均为类石墨相结构. 3 000~3 500 cm-1之间的宽峰为N—H伸缩振动[36],表明存在未缩合的氨基官能团;1 200~1 650 cm-1区域的峰对应CN杂环的不对称伸缩振动[37];1 645、1 573、1 466 和 1 418 cm-1出峰对应衍生自七嗪环的重复单元的伸缩振动,与已有研究结果[37-38]一致;1 328 和 1 250 cm-1特征峰为七嗪环的弯曲振动[37];880 cm-1处的吸收带为氨基不完全缩合引起的NH变形;810 cm-1出峰为三嗪环的典型径向振动. 综上,TCN的基本组成单元仍为以七嗪环为主的石墨相氮化碳.
GCN与TCN的XRD谱图〔见图1(b)〕显示:GCN的XRD谱图在27.4°与13.0°存在2个特征峰,分别对应g-C3N4的(002)晶面与(100)晶面,与已有研究结果[1,26,35]一致;TCN的XRD谱图中同样存在(002)晶面与(100)晶面所对应的特征峰,但(100)晶面特征峰由13.0°偏移至12.8°,证明TCN的分子结构中存在更多、更有序的七嗪环[35],七嗪环较三嗪环结构更稳定,便于电子转移. 此外,TCN(100)晶面特征峰出峰强度明显强于GCN,证明TCN中的层状结构明显,堆叠层数更多. 由于七嗪环边缘的N原子含有孤对电子,增强的(100)晶面将暴露更多七嗪环边缘的孤对电子,利于光生电子激发和载流子分离,从而促进光催化反应. 此外,TCN结构利于共轭体系形成,使该区域的电子密度增大,降低光激发后电子跃迁至导带所需能量[35,39];同时,TCN的(002)晶面特征峰由27.4°偏移至27.5°,证明TCN的层间距缩小[40],利于电子转移与传质等过程.
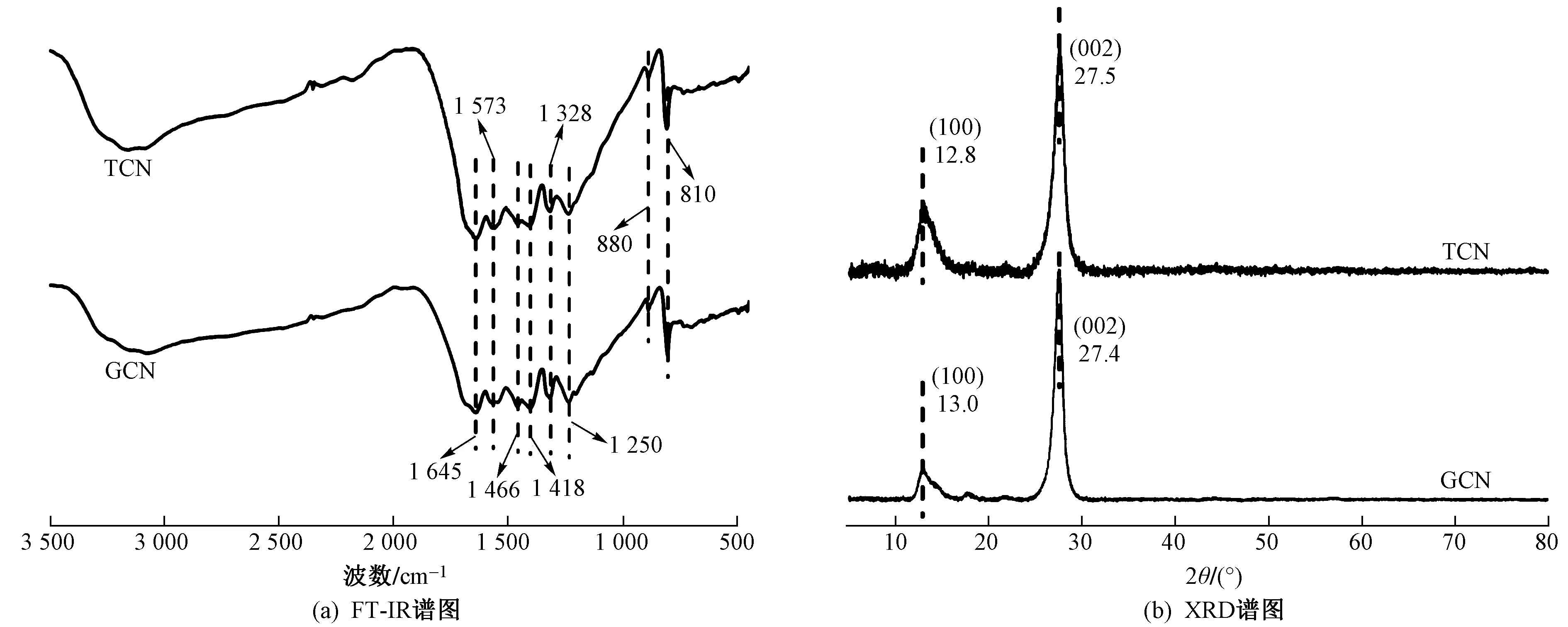
图1 GCN与TCN的FT-IR谱图与XRD谱图Fig.1 FT-IR spectra and XRD pattern of GCN and TCN
图2为GCN与TCN的SEM表征结果. 由图2可见:GCN呈超薄片层无序堆叠形成的块状形态[1,41];TCN呈管状结构,由二维超薄片层有序层叠而成,长约140 μm〔见图2(b)〕. TCN的管状结构呈端口开口状态,中空腔体直径约为5 μm〔见图2(c)〕,且TCN管截面呈八边形柱状结构〔见图2(d)〕.该形貌结构使材料比表面积增加,并通过管状限域效应(通过使污染物富集在反应活性位点上与调节电子结构等行为提升反应速率[42-45]),实现污染物降解率的提升. TCN的BET比表面积为20.9 m2/g,大于GCN (8~10 m2/g)[26,32,37],TCN比表面积的提升利于TCN表面暴露更多的活性位点进行光催化反应. 而TCN的BJH孔径分布曲线与已有研究中体相GCN的孔径分布[26,37-38]相似,即主要以孔径小于2 nm的微孔与孔径在2~50 nm之间的中孔组成.

图2 GCN与TCN的SEM图Fig.2 SEM images of GCN and TCN
图3为TCN与GCN的紫外-可见漫反射光谱. 由图3(a)可见,与GCN相比,TCN表现出明显的红移,证明TCN拥有更窄的带隙,且在全波段范围内表现出更强的光吸收率,特别是在可见光范围内. 光催化反应中光生空穴/电子的氧化/还原能力与光催化剂的带隙直接相关,基于Kubelka-Munk理论〔见式(1)〕对UV-vis DRS数据进行转化[27],从而计算能带带隙值(Eg). 由图3(b)可见,GCN的Eg为2.69 eV,与已有研究结果(2.70 eV)[1,26,37]接近;而TCN的Eg为2.48 eV,明显小于GCN.
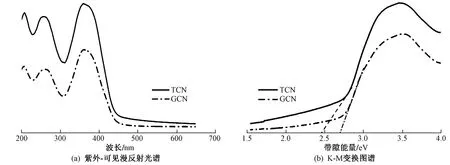
图3 TCN与GCN的紫外-可见漫反射光谱Fig.3 UV-vis DRS spectra of TCN and GCN
αhv=A(hv-Eg)n/2
(1)
式中:α为吸收系数;h为普朗克常数;v为光的频率,Hz;A为比例常数;Eg为带隙能量值,eV;n为基于材料特性的系数. 当材料为间接带隙型半导体,n取1;当材料为直接带隙型半导体,n取4;该研究中的材料均为直接带隙型半导体,故n取4.
2.2 光电化学性能测试
图4为TCN与GCN两种材料光电化学测试结果. 图4(a)为空间电荷区内的莫特-肖特基曲线,根据莫特-肖特基公式〔见式(2)〕,可计算出材料平带电位.
(2)
式中:C为界面电容,μF/cm2;ε为相对介电常数;ε0为真空介电常数,取8.85×10-14F/cm;V为外加电位,V vs. NHE;VFB为平带电位,V vs. NHE;e为带电量,取1.60×10-19C;kb为玻尔兹曼常数,取1.38×10-23J/K;T为温度,取298 K. 由于该体系中的界面势垒(kbT)小于外加电压约2个数量级,故界面势垒可忽略不计.
由式(1)可知,催化剂的平带电位与界面电容平方的倒数成正比. 由于该研究使用的参比电极为Ag/AgCl,最终根据式(1)拟合得到GCN与TCN的平带电位分别为-1.02与-0.80 V vs. NHE. 由于导带电势(ECB)比平带电位低约0.1 V vs. NHE[27,46],故GCN与TCN的导带电势的绝对位置分别为-1.12与-0.90 V vs. NHE. 已有研究[29,35,46]发现,GCN的导带电势绝对位置为-0.96~-1.20 V vs. NHE,证明该研究中导带电势绝对位置的估算方法较可靠. 根据催化剂的导带电势的绝对位置与能带带隙值,推算出GCN与TCN价带电势的绝对位置分别为1.57与1.58 V vs. NHE,证明TCN实现了有效的能带结构调控,且调控主要发生在材料的导带电位上. 由图4(b)可见,TCN拥有更小的电阻与更高效的界面电荷转移效率. 图4(c)为GCN与TCN的光电流测试(PEC)结果,显示TCN具有更强的光生载流子的产生能力与迁移能力,TCN的光电流没有明显衰减,证明TCN比GCN更加稳定. 线性扫描伏安测试结果〔见图4(d)〕显示,TCN拥有更低的初始电压与更高的电流密度. 综上,TCN的构型显著降低了电荷转移的势垒,提升了电荷转移效率,抑制光生空穴与电子的复合[47].
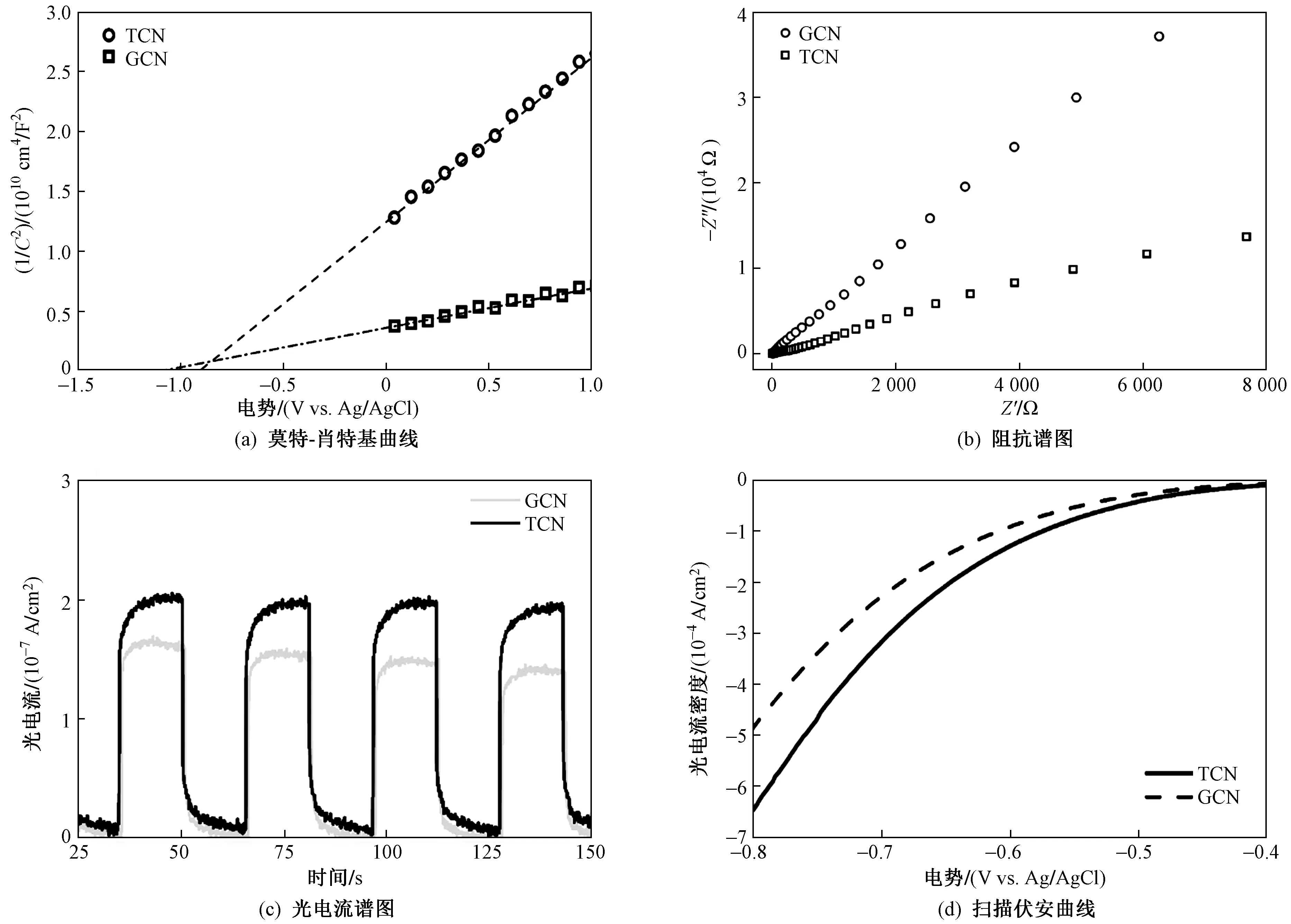
图4 TCN与GCN的莫特-肖特基曲线、阻抗谱图、光电流谱图和扫描伏安曲线Fig.4 Mott-Schottky plot, EIS, photocurrent response, and LSV of TCN and GCN
2.3 DCF光催化降解试验
图5(a)为DCF的光催化降解动力学结果. 引入Langmuir-Hinshelwood (L-H)模型简化所得的准一级动力学模型对动力学过程进行拟合[16,23],该模型表示为
ln(Ct/C0)=-k1t
(3)
式中:C0与Ct分别为DCF光催化降解反应初始时刻与反应过程中t时刻的DCF浓度,mg/L;k1为准一级动力学常数,min-1.
预试验结果显示,暗室条件下DCF在30 min达到吸附-解吸平衡,吸附量小于2%,可忽略不计. 此外,在未加入催化剂时,DCF的90 min光解去除率小于1%. GCN在DCF去除过程中表现出较低的光催化活性,90 min去除率仅为61.0%,且矿化率为21.8%(见表1). 而TCN表现出更高的DCF光催化降解效率,在60 min可降解96.8%的DCF,并在90 min彻底降解DCF,矿化率较高(51.9%). 此外,TCN光催化降解DCF的k1值为6.99×10-2min-1,是GCN (k1=1.28×10-2min-1)的5.5倍. DCF的快速降解可归因为TCN具有更强的可见光利用率、更高的载流子分离效率以及被抑制的光生空穴-光生电子复合速率. 为探究溶液pH对DCF降解的影响,该研究在不同pH(5、7和9)下进行DCF降解试验〔见图5(b)〕. 由图5(b)可见:碱性条件有利于DCF的降解. pH为9时,TCN在60 min实现DCF的100%降解;但pH为5时,TCN在60 min只降解约90%的DCF. pH对GCN降解DCF的影响与TCN降解DCF的结果相似,即碱性条件促进DCF降解.
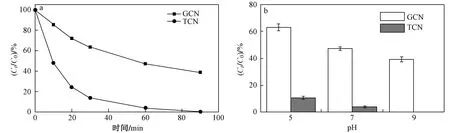
图5 DCF降解动力学与pH对60 min内DCF光催化降解速率的影响Fig.5 Photodegradation kinetic of DCF and pH effects on DCF photodegradation at 60 min

表1 GCN与TCN降解DCF的矿化率与动力学拟合结果
2.4 DCF光催化降解机理
为探究DCF的降解机理,需鉴定参与DCF降解的活性物种. 光催化反应中常见的活性物种包括羟基自由基(·OH)、光生空穴(h+)和超氧根自由基(·O2-)等. 光催化材料(如GCN和TCN)受自然光照射后,能量大于或等于能隙的部分被材料吸收,光子激发价带上的电子跃迁至导带形成导带上光生电子,同时在VB上留下光生空穴. 而产生的光生空穴-电子对(h+-e-)又易发生复合而耗散能量. 导带上光生电子可与水中的溶解氧反应,形成超氧根自由基(·O2-);价带上光生空穴可氧化水或OH-形成羟基自由基. 该研究中GCN和TCN导带电势的绝对位置分别为-1.12与-0.90 V vs. NHE,均高于O2/·O2-的氧化/还原电位(-0.33 V vs. NHE)[27,48],故该研究体系中存在大量·O2-. 但GCN和TCN价带电势的绝对位置分别为1.57与1.58 V vs. NHE,均低于H2O/·OH或OH-/·OH 的氧化/还原电位(分别为2.73与1.99 V vs. NHE)[1,16,49],故空穴不能直接氧化H2O或OH-生成·OH.
图6(a)为·O2-的电子自旋共振图谱. 由图6(a)可见,GCN与TCN光催化反应均可产生·O2-,且TCN体系中·O2-的信号强度远高于GCN,表明TCN具更优异的光催化性能. ·OH电子自旋共振谱图中,测试结果与基线无显著差异,故没有羟基自由基参与反应. 图6(b)为活性物种淬灭试验结果. 由图6(b)可见,当引入淬灭剂KI、IPA、BQ后,GCN体系中DCF的1 h降解率从52.3%分别降至24.4%、50.5%与10.6%,而TCN体系中DCF的1 h降解率从96.8%分别降至76.6%、95.2%与28.6%. 因此,在该光催化体系中·O2-为主导反应的活性物种,与电子自旋共振结论一致,同时光生空穴的直接氧化也对DCF光催化降解有贡献. 此外,碱性条件下,大量OH-将吸附在催化剂表面,催化剂表面电负性增强,进一步通过能带调控机制增强TCN光催化材料的催化活性,产生更多光生电子,最终促进关键活性物种·O2-的产生[1,27]. 另外,酸性条件下·O2-(E0=1.83 V vs. NHE)[16]会被质子化形成·OOH(E0=1.48 V vs. NHE)[50],进而降低氧化能力,降低DCF的降解速率[1]. 综上,DCF的光催化降解及完全矿化过程如式(4)~(8)所示.
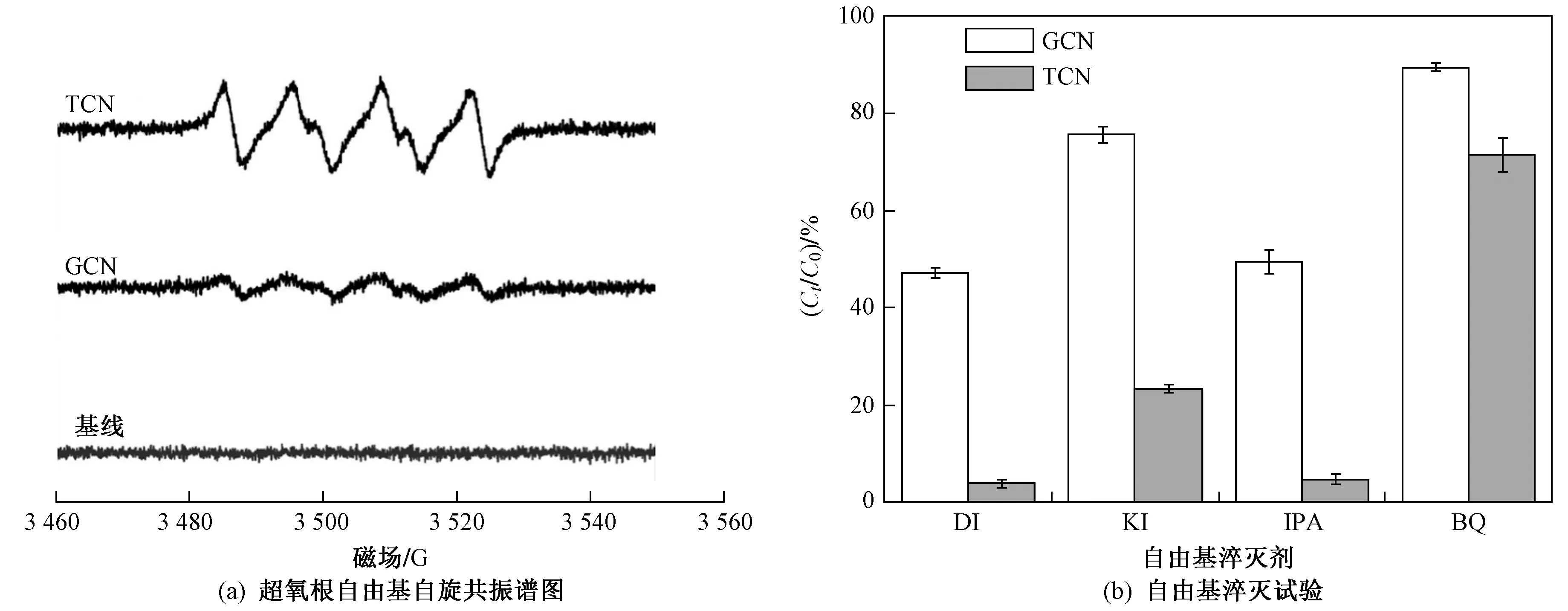
图6 TCN与GCN超氧根自由基的电子自旋共振谱图与淬灭试验Fig.6 ESR spectra of TCN and GCN for superoxide radical and scavenger effect on DCF degradation
GCN/TCN+h→GCN(e--h+)/TCN(e--h+)(4)
GCN-e-/TCN-e-→GCN/TCNe-
(5)
O2+e-→·O2-
(6)
·O2-+H+→HOO·
(7)
·O2-/·OOH/h++DCF→CO2+H2O+Cl-
(8)
2.5 TCN重复利用
由图7可见,在TCN 5次循环利用过程中,反应1 h 后DCF降解率没有显著下降,均维持在92%之上. 结果表明,该研究所制备的TCN光催化剂较稳定,具有实际应用前景.
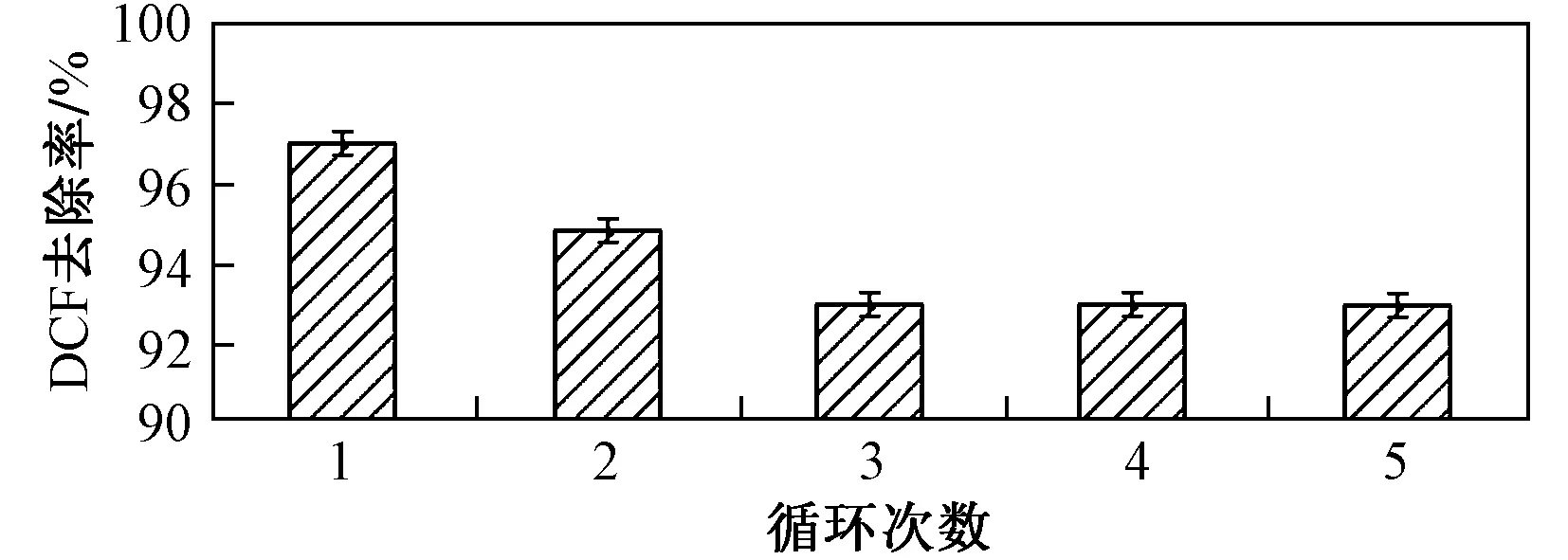
图7 TCN降解DCF的循环利用试验Fig.7 Reusability test of DCF photodegradation by TCN
3 结论
a) 该研究通过使用超分子自组装前驱体进行热聚合反应,合成了一种呈管状的石墨相氮化碳(TCN)材料,该催化剂具备比表面积大、结构单元有序排列、光稳定性强等特点.
b) TCN在模拟太阳光下可高效、快速光催化降解水中新污染物DCF,其降解反应的准一级动力学常数是传统石墨相氮化碳的5.5倍.
c) TCN光催化性能提升有4个主要原因:① TCN 的比表面积较大,拥有更多的表面活性位点;②晶面调控使(100)晶面暴露增强,在光催化过程中暴露出更多的孤对电子,使电子易受光激发后从价带跃迁至导带;③ TCN能带间隙更窄,可见光吸收提升;④电子传导能力增强,提升载流子分离效率,抑制光生空穴与光生电子复合受.
d) 电子自旋共振谱图证实,在太阳光光催化反应中TCN较GCN能产生更多的超氧根自由基,超氧根自由基为主导DCF光催化降解反应的活性物种;同时,光生空穴也对DCF光催化降解有贡献.
e) 该研究提供了一种采用超分子自组装合成大比表面积和窄带氮化碳催化材料的方法,所制备的新型光催化材料可为水中新污染物,尤其是活性药物的去除提供可参照的方法.
