高寒湿地土壤微生物多样性对氮沉降浓度差异的响应
2021-12-20徐润宏朱锦福刘泽华
徐润宏, 谭 梅, 朱锦福, 刘泽华
(1. 青海师范大学 生命科学学院 青海省青藏高原药用动植物资源重点实验室,西宁 810008; 2. 青海师范大学 生命科学学院 青海省青藏高原生物多样性形成机制与综合利用重点实验室,西宁 810008)
氮是陆地生态系统的限制因素之一。大气氮沉降的急剧增加给陆地生态系统造成的不良影响已引起相关学者的普遍关注,如土壤碳氮含量变化,植物生物量及多样性变化,微生物分解等。
土壤微生物是土壤中最活跃和最丰富的生物类群[1],是陆地生态系统养分循环的重要参与者,在土壤氮循环中起重要作用[2]。研究表明,短期模拟氮沉降条件会使微生物量增加的原因是土壤有效养分含量增加[3]。氮沉降改变了土壤微生物生物量、真菌和细菌的比值,从而使群落结构发生改变[4]。也有研究表明,氮沉降对土壤微生物的生长有抑制作用[5],但其变化规律还不明了,需要进一步了解。
青藏高原是全球变化的敏感区和生态脆弱区,在全球生态系统中居于重要生态地理位置[6]。在生态系统碳氮循环中发挥重要作用[7-8]。青海湖位于青藏高原东北部,是青海省东北部重要的生态安全屏障,在维持青藏高原和我国西北部生态安全平衡等方面具有重要作用[9]。氮沉降背景下有关高寒湿地土壤微生物的研究还较少,揭示其对氮沉降的响应规律有助于更好地了解氮沉降对高寒湿地生态系统的影响,并对生态系统功能变化提供参考。基于此,本文通过不同水平氮添加试验,利用高通量测序技术测定细菌和真菌的群落组成,为青藏高原湿地生态系统安全及管理提供理论参考。
1 材料与方法
1.1 研究区概况
研究区位于青藏高原东北部青海湖流域小泊湖观测站(36.78° N, 100.80° E),青海湖湖东,距离湖东种羊场约6 km,是青海湖水位下降遗留下来的沼泽化草甸湿地,平均海拔约3 200 m。该地具有典型的高原大陆性气候,光照充足,雨量较少,降水主要集中于6月—9月。主要植物类群为华扁穗草 (Blysmussinocompressus) 、禾叶嵩草 (Kobresiagraminifolia)和藏嵩草(Kobresiatibetica) 等[10]。
1.2 试验设计及样品采集
共设置N2(2 g/m2)、N5(5 g/m2)和N10(10 g/m2)共3个氮添加处理和N0(0 g/m2)1个空白对照处理试验,每个处理设置3个重复,一共分12个1 m×1 m样方,样方间隔1 m作为缓冲带。施加氮源为NH4NO3,按不同施氮梯度,称取相应的NH4NO3,溶于500 mL水中,用喷壶喷洒于相应样方中,对照喷洒等量的水。从2019年6月至8月,每月中旬取样后施药。
于2019年8月中旬植物生长旺盛时采样,在每块样方中按照五点取样法进行采样,采用螺旋取土钻,按照0~15 cm和15~30 cm两层进行取样,尽可能去除植物根系及凋落物,放入无菌自封袋中,将约400 g土样迅速带回实验室,并放入冰箱4 ℃冷藏保存,用于测定土壤微生物。
1.3 土壤微生物测定
1.3.1 微生物多样性提取试剂盒
使用MN NucleoSpin 96 Soil试剂盒/PowerSoil DNA Isolation kit强力土壤DNA提取试剂盒。
1.3.2 扩增引物
细菌16S rRNA (V3+V4)区域引物:正向引物5′- ACTCCTACGGGAGGCAGCA-3′;反向引物5′- GGACTACHVGGGTWTCTAAT-3′。真菌ITS1 区域引物:正向引物5′-CTTGGTCATTTAGAGGAAGTAA-3′;反向引物5′-GCTGCGTTCTTCATCGATGC-3′。
1.3.3 PCR扩增
目标区域PCR(10 μL体系):基因组DNA 50 ng±20%,Vn F(10 μmol/L)0.3 μL,Vn R(10 μmol/L)0.3 μL,KOD FX Neo Buffer 5 μL,dNTP (2 mmol/L each) 2 μL,KOD FX Neo 0.2 μL,ddH2O补至10 μL。反应条件:95 ℃预变性5 min;95 ℃变性30 s,50 ℃退火30 s,72 ℃延伸40 s,20个循环;72 ℃再延伸7 min;4 ℃ ∞。Solexa PCR体系:目的区域PCR 纯化产物5 μL,MPPI-a(2 μmol/L)2.5 μL,MPPI-b(2 μmol/L)2.5 μL,2×Q5 HF Mmol/L 10 μL。反应条件: 98 ℃预变性30 s;98 ℃变性10 s,65 ℃退火30 s,72 ℃延伸30 s,10个循环;72 ℃再延伸5 min。1.8%的琼脂糖凝胶。电压120 V,40 min。将得到的PCR产物根据电泳定量结果,按质量比1∶1进行混样。混样后采用OMEGA DNA纯化柱进行过柱纯化。1.8%的琼脂糖凝胶,120 V、40 min 电泳后切目的片段,并回收。基于 Illumina HiSeq测序平台,对纯化的样品进行测序分析。
1.4 数据分析
使用Usearch软件对Tags在97%的相似度水平下进行聚类、获得OTU,并基于Silva(细菌)和UNITE(真菌)分类学数据库对OTU进行分类学注释,进而在各水平统计各样品群落组成。利用QIIME软件生成不同分类水平上的物种丰度表,再利用R语言工具绘制成样品各分类学水平下的群落结构图。Mothur(version v.1.30)软件分析样品的α多样性(Alpha diversity),R语言绘制样品 β 多样性(Beta diversity)分析图。接着用SPSS 20.0对数据进行统计分析,采用单因素方差分析(One Way ANOVA)和LSD(最小显著差异法)比较各处理水平之间的差异。
2 结果与分析
2.1 稀释性曲线
从细菌和真菌在各处理梯度下的稀释性曲线(图1)可以看出,在当前样本量下,曲线趋于平缓,OTU数目不再随着样本量的增加而大量增加,说明当前测序样本量已足够,测序结果合理,当前测序深度可反应本次土壤细菌、真菌的真实情况。
2.2 细菌、真菌的Venn图分析
通过聚类得到各样品独有和共有的OTU个数(图2)。细菌通过聚类,总共得到1 599个OTU,N0、N2、N5和N10处理分别得到1 528、1 544、1 547和1 548个OTU。其中,有1 508个OTU为4种处理下共有的,均达到各处理梯度下总OTU的95%以上,N0和N5独有的OUT数为2,N5和N10没有独有的OUT。真菌通过聚类,总共得到1 312个OUT,N0、N2、N5和N10处理分别得到915、879、701和786个OTU。其中,所有处理共有的OTU数目为345,分别占据N0、N2、N5和N10处理的37.70%、39.25%、49.22%和43.89%,各处理独有的OTU数分别为122、60、37和99。
2.3 微生物群落结构分析
在门类水平上分别选取细菌和真菌相对丰度为1%的类群,并将其他物种合并为 Others 在图3中显示,Unclassified代表未得到分类学注释的物种。细菌相对丰度1%的物种有变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、芽单胞菌门(Gemmatimonadetes)、拟杆菌门(Bacteroidetes)、己科河菌门(Rokubacteria)、Patescibacteria、硝化螺旋菌门(Nitrospirae)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)、浮霉菌门(Planctomycetes)、Latescibacteria、Nitrospinae、螺旋菌门(Spirochaetes)和蓝藻菌门(Cyanobacteria)共16种,占据了整个细菌群落的98%以上,其中变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)和放线菌门(Actinobacteria)相对丰度均在10%以上,总计占据整个细菌群落的75%以上,为主要优势物种。变形菌门(Proteobacteria)在两土层中的相对丰度随施氮浓度的上升均呈现降低趋势,且N2处理显著低于N0处理(P<0.05),N5和N10处理达到极显著水平(P<0.01);绿弯菌门(Chloroflexi)相对丰度在0~15 cm土层中随施氮浓度的增加而降低但变化不显著(P>0.05),在15~30 cm土层中也呈现降低趋势,且在N10处理下显著低于N0处理(P<0.05);酸杆菌门(Acidobacteria)的相对丰度与变形菌门(Proteobacteria)的趋势相反,在两土层中均随施氮浓度的上升逐渐增加,且都在各处理下均达到显著水平(P<0.05);放线菌门(Actinobacteria)在两土层中的相对丰度在氮处理下均低于N0处理,在0~15 cm土层中没有达到显著水平(P>0.05),在15~30 cm中N10处理显著低于N0处理(P<0.05)。真菌主要优势菌门有子囊菌门(Ascomycota)、担子菌门(Basidiomycota)和被孢菌门(Mortierellomycota),相对丰度大于1%的还有球囊菌门(Glomeromycota)和壶菌门(Chytridiomycota)。子囊菌门(Ascomycota)的相对丰度在0~15 cm土层中随施氮浓度的增加呈现先降低后增加趋势,N2处理下达到最低但不显著(P>0.05),N10处理下达到最高且显著高于N0处理(P<0.05),15~30 cm土层中,其相对丰度随施氮浓度增加成逐渐上升趋势,在N10处理下达到显著水平(P<0.05);0~15 cm土层中担子菌门(Basidiomycota)的相对丰度在各处理下均低于N0处理,但差异不显著(P>0.05),15~30 cm土层中,其相对丰度随施氮浓度逐渐增加,N10处理显著高于N0处理(P<0.05);0~15 cm土层中被孢菌门(Mortierellomycota)的相对丰度在N5和N10处理下显著高于N0处理(P<0.05),在15~30 cm土层中的相对丰度逐渐降低,但变化不显著(P>0.05)。可知,氮沉降背景下,微生物菌群的结构发生了显著改变,但是细菌和真菌有所差别,且不同土层中的变化也有一定的差异。

图3 细菌(a)和真菌(b)优势菌门变化Figure 3 Changes of dominant phylum of bacteria(a)and fungi(b)

图中横坐标为不同氮处理。图4 细菌在0~15 cm土层(a)和15~30 cm土层(b)的α多样性指数Figure 4 The diversity index of bacteria at layers soil of 0-15 cm(a)and 15-30 cm(b)
2.4 微生物α多样性分析
Chao1指数衡量物种丰富度即物种数量的多少,Simpson指数衡量物种优势度,Shannon指数表示物种多样性。0~15 cm土层中细菌的丰富度和优势度指数在施氮条件下均呈现下降趋势,但差异不显著(P>0.05);多样性指数随施氮浓度的上升逐渐增加,且N10处理显著高于N0处理(P<0.05)。15~30 cm土层中细菌的丰富度指数和优势度指数随施氮浓度均呈现先降低后增高的趋势,而多样性指数呈现相反的趋势,差异都没有达到显著水平(P>0.05),具体见图4。0~15 cm土层中真菌的丰富度指数在氮处理下减少,其中N5处理达到最低,极显著低于N0处理(P<0.01);优势度指数呈先增加后减少的趋势,在N2处理达到最高值,N10处理达到最低值,且N10处理显著低于N0处理(P<0.05);多样性指数增加,且N10处理显著高于与N0处理(P<0.05)。15~30 cm土层中,真菌的丰富度指数和多样性指数随施氮浓度的增加呈先减少后增加的趋势,而优势度指数呈先增加后减少的趋势,且均在N5处理下达到最低和最高值,但差异均不显著(P>0.05),具体见图5。
2.5 施氮条件下细菌和真菌群落的差异
非度量多维标定法(NMDS)的Stress小于0.2时,表明分析具有一定的可靠性,在坐标图上距离越近的样品,相似性越高。基于Bray-curtis距离算法得出细菌和真菌的NMDS分析(图6),分析结果的Stress值均小于0.2,表示NMDS分析结果合理。非参数多元方差分析(Permanova)显示,细菌和真菌群落的P值均小于0.01,细菌和真菌的群落结构在施氮条件下均发生了显著改变。
UPGMA聚类树显示,土壤细菌群落总体在N0、N2和N10处理下的组成较相近,距离N5水平较远,土壤真菌群落N0与N2处理下的组成较相近,其次是N5处理,距离N10处理下的土壤真菌群落最远。随着施氮浓度的上升,土壤微生物物种组成差距加大。
为进一步鉴别不同处理的显著差异和主要贡献生物类别,采用微生物组间差异分析(Lefse)检验不同处理间的差异贡献关系。结果(图7)表明,真菌在N0、N5和N10处理下均有差异显著的生物标记类群,N0处理下门水平的显著标志类群是变形菌门(Proteobacteria),α-变形杆菌纲(Alphaproteobacteria)是纲水平上的显著标志物种,目水平上有红螺菌目(Rhodospirillales)和Microtrichales,科水平上有红螺菌科(Rhodopirillaceae)和Ilumatobacteraceae,属水平上有Defluviicoccus,种水平上是uncultured_bacterium_g_Defluviicoccus。N5处理下门、纲、目、科、属、种水平下均有显著贡献类群,分别是酸杆菌们(Acidobacteria)、Blastocatellia_Subgroup_4、Pyrinomonadales、Pyrinomonadaceae、RB41、uncultured_bacterium_g_RB41。N10处理下门水平上显著贡献类群是芽单胞菌门(Gemmatimonadetes),科水平上是MND1,uncultured_bacterium_g_MND1是种水平上的贡献类群。真菌在N0和N10处理下有显著贡献物种,N0处理下属水平上是Neoascochyta。N10处理下门水平上的贡献类群是子囊菌门(Ascomycota),纲水平上是散囊菌纲(Eurotiomycetes)和子囊菌纲(Sordariomycetes),目水平上有煤炱目(Capnodiales)和(肉座菌目)Hypocreales,赤壳科(Nectriaceae)和毛壳菌科(Chaetomiaceae)是科水平上的显著贡献类群。

图中横坐标为不同氮处理。图5 真菌在0~15 cm土层(a)和15~30 cm土层(b)的α多样性指数Figure 5 The diversity index of fungi in layers soil of 0-15 cm(a)and 15-30 cm(b)

图6 细菌(a)和真菌(b)的NMDS分析Figure 6 NMDS analysis of bacteria and fungi
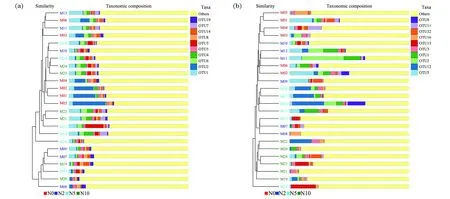
图7 土壤真菌(a)和细菌(b)聚类树柱状图组合图Figure 7 UPGMA combined with histogram chart of fungi(a)and bacteria(b)
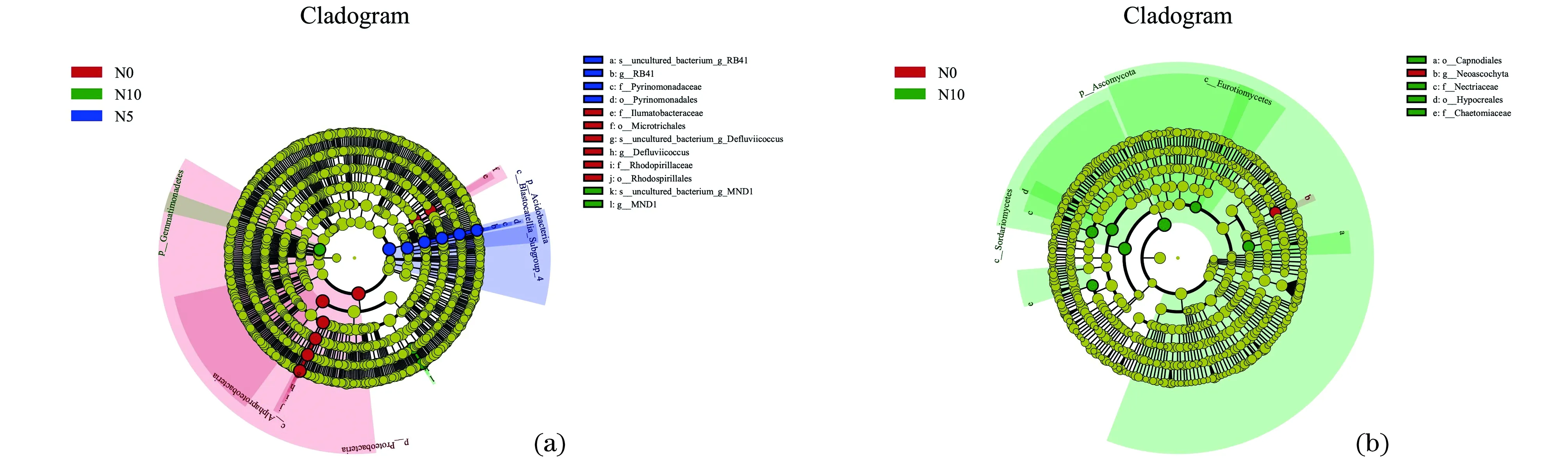
图8 Lefse分析土壤细菌(a)和真菌(b)的差异显著标志类群Figure 8 Lefse analyzed significant marker groups of differences between soil bacteria(a)and fungi(b)
3 讨论与结论
大气氮沉降是目前全球变化中的热点问题之一,可直接或间接影响生态系统的结构与功能。土壤微生物是土壤中重要组分之一,是影响土壤健康评价的关键因子[11]。研究表明:土壤细菌的优势菌门有变形菌门、放线菌门、绿弯菌门、酸杆菌门、厚壁菌门、疣微菌门和芽胞菌门[12];真菌优势菌门有子囊菌门、担子菌门、被孢菌门和壶菌门[13-14]。本研究发现,变形菌门、绿弯菌门、酸杆菌门、放线菌门、子囊菌门、担子菌门和被孢菌门为优势菌门,与上述研究基本一致。但氮添加改变了微生物群落各门类的相对丰度,如变形菌门和放线菌门在各氮处理下相对丰度均低于N0对照处理,变形菌门在N2处理显著低于N0处理,N5和N10更是达到了极显著水平;放线菌门的相对丰度在N10处理下也显著低于N0处理;酸杆菌门的相对丰度在施氮处理下显著高于N0处理。研究认为,酸杆菌门与土壤氮含量有正相关关系[15],施氮使土壤氮含量增加,从而使酸杆菌门的相对丰度增加。Fierer等[16]认为,当土壤中氮素增加时,一些富营养型的菌群丰度会增加,如变形菌门等。本研究变形菌门的相对丰度随氮处理呈下降趋势,这可能与高寒湿地生态系统的脆弱性和敏感性有关,但具体机理还有待进一步研究。本研究中子囊菌门的相对丰度为43.34%~68.15%,子囊菌门占据土壤菌群的主要地位,且在氮处理下均高于N0处理。这可能与其强大的环境适应性有关。担子菌门相对丰度与土壤氮呈负相关关系[17],担子菌门的相对丰度在0 ~15 cm土层中减少,在15~30 cm土层中增加,可能是施氮主要影响上层土壤氮含量,使上层土壤担子菌相对丰度下降,下层变化相反可能是因为土壤的富营养状态,使富营养型菌群增加,降低菌群竞争从而使担子菌门相对丰度有所上升。
氮沉降会改变土壤微生物的群落结构和多样性[18-19]。研究表明氮添加改变了土壤微生物群落结构[20]。由细菌和真菌的Venn图(图2)可知,各处理下细菌OTU数均达到了总OTU数的95%以上,各处理下真菌的OTU数占据总OTU数的37.70%~49.22% ,表明细菌群落相较于真菌群落总体对氮沉降有较强的稳定性,真菌群落结构变化大于细菌。NMDS分析(图6)表明,在不同的氮处理水平下,土壤微生物群落发生了显著改变。UPGMA聚类树(图7)也显示,氮处理下,土壤细菌和真菌的群落组成产生了显著变化。微生物优势菌门的结构分析中发现,在N2处理中,放线菌门和酸杆菌门的相对丰度与对照处理产生显著差异,而子囊菌门的相对丰度在高浓度氮N10处理中与对照产生显著差异,表明细菌较真菌对氮沉降更为敏感。真菌拥有极强的纤维素降解能力[25],在较小范围的环境变化下有更好的调节能力及稳定性。土壤细菌的丰富度指数和优势度指数在两土层中的变化均不显著,主要表现为下降,多样性指数在0~15 cm土层中有显著增加(图4)。分析认为,可能是氮添加使微生物活性受抑制,减少了优势物种的出现,降低了土壤细菌的竞争,从而使细菌多样性上升[22]。氮处理使土壤真菌的丰富度指数和多样性指数显著下降而优势度指数增加(图5),真菌受环境影响较大,真菌群落对不断增加的氮处理有弹性响应[23]。真菌群落在不同氮处理水平下的结构差异可能与试验地环境、海拔高度、施氮时间等多种因素相关[24]。
氮沉降背景下,土壤细菌和真菌的多样性与群落结构均发生显著变化,但响应程度不相同,细菌较真菌对氮沉降的感应敏感。本研究在短期氮沉降背景下探讨了土壤微生物群落的分布差异,可为短期氮沉降背景下的环境响应提供参考,却无法深入评估长期氮沉降变化下湿地土壤环境的变化过程。今后应注重长期持续的观测实验,来揭示生态系统对氮沉降的具体响应机制。
