不同血清型视神经脊髓炎谱系疾病双向队列研究
2021-12-14滕新岭常旭婷李尚茹包新华张月华姜玉武
滕新岭 常旭婷 张 捷 李尚茹 武 元 谢 涵 包新华 张月华 姜玉武 吴 晔
视神经脊髓炎(NMO)既往被认为是多发性硬化的变异型,2007年视神经脊髓炎谱系疾病(NMOSD)的概念被提出[1]。NMOSD属于中枢神经系统特发性炎症性脱髓鞘疾病,主要累及视神经和脊髓,也可累及延髓最后区、导水管周围、下丘脑、皮质下和脑室周围白质、胼胝体及深部灰质。水通道蛋白4(AQP4)是星形胶质细胞表达的特异性抗原,其在NMOSD患者血清中被发现,且被认为是与NMOSD发病密切相关的特异性生物标记物。2015年国际NMO诊断小组(IPND)制定了新的NMOSD诊断标准,根据血清学分为AQP4-IgG阳性和AQP4-IgG阴性/未知2类[2]。近5年来,随着血清抗髓鞘少突胶质细胞糖蛋白IgG(MOG-IgG)检测的可及性及其相关疾病的认识,在AQP4-IgG阴性/未知的一部分NMOSD患者中发现血清MOG-IgG阳性。因此可以将NMOSD分为AQP4-IgG阳性、MOG-IgG阳性和血清学阴性(2种抗体均阴性)3类。目前对于不同血清型NMOSD患儿的临床表型差异尚不明确。本研究通过临床随访比较不同血清型NMOSD患儿的临床表型差异,以利于临床识别。
1 方法
1.1 研究设计 动态双向队列研究。2012年1月至2021年3月在北京大学第一医院(我院)儿科诊断为NMOSD患儿即进入队列,回顾性采集既往的临床资料,随访观察病例临床特征,队列终点为起病至末次随访时间≥6个月。
1.2 伦理及知情同意 本研究经我院生物医学研究伦理委员会批准,批准文号:(2018)科研第(98)号。患儿家长均签署书面知情同意书。
1.3 诊断标准 中枢神经系统炎症性脱髓鞘疾病患儿中同时符合以下标准:①起病年龄<18岁。②符合IPND提出的NMOSD诊断标准[2][包括6个核心临床特征:视神经炎(ON)、急性脊髓炎、延髓最后区综合征、脑干综合征、间脑综合征和大脑综合征]。若AQP4-IgG阳性,则需≥1项核心临床特征,排除其他疾病即可诊断;若AQP4-IgG和MOG-IgG双阴性,需≥2项核心临床特征,其中的1项为视神经炎、或急性脊髓炎、或延髓最后区综合征,且符合MR附加条件[2],排除其他疾病才能诊断。
1.4 纳入标准 ①2012年1月至2021年3月我院儿科诊断为NMOSD的患儿;②起病至末次随访时间≥6个月。
1.5 数据收集及随访 建立队列临床观察量表。(1)回顾性采集信息,首次在我院诊断NMOSD的患儿收集发病以来的临床症状和体征,在他院已经诊断的NMOSD患儿收集既往的诊疗经过。(2)临床随访方式及信息收集,①无复发病例:要求每半年来院或接受我院电话随访1次,根据症状体征进行必要的检查和调整治疗方案,②复发病例:要求即来院复诊,根据症状体征进行必要的检查。收集随访期间的临床症状、体征、影像学和实验室检查资料。
1.5.1 基线情况 起病年龄,性别,起病至末次随访病程,总发作(新出现的持续时间>24 h的中枢神经系统症状和体征,除外其他原因引起的临床或影像学改变)次数。

1.5.3 扩展残疾状态量表(EDSS)评分 每次来我院随访行EDSS评分。①功能系统评分(FS):视觉功能 (0~6分)、脑干功能(0~5分)、锥体功能 (0~6分)、小脑功能(0~5分)、感觉功能(0~6分)、直肠和膀胱功能(0~6分)、大脑功能(0~6分),②行走功能(0~12分),③总分(0~10分)。0分为神经检查正常;1分为没有残疾,只有1个FS轻度异常体征;9分为卧床不起,可以交流,吃饭;10分为死亡。以最后一次随访EDSS评分纳入本文分析。
1.5.4 头颅MR分类 在我院和他院的每次发作急性期的头颅MR,新病灶累及的部位包括10类:皮层、皮层下白质、脑室旁白质、胼胝体、内囊、丘脑、基底节、脑干、导水管/四脑室周围、小脑。
1.5.5 脊髓MR新病灶部位分段 在我院和他院的脊髓MR检查,分为颈段、胸段和腰骶段3类,评估病灶是否为长节段脊髓炎(LETM),即病灶≥3个连续椎体节段[4]。
1.5.6 实验室检查 包括在我院和他院的实验室检查结果,记录每次血清MOG-IgG、AQP4-IgG检测时间和滴度;急性期脑脊液常规和生化;血清及脑脊液抗N-甲基-D-天冬氨酸受体(NMDAR)抗体、甲状腺抗体和脑脊液特异性寡克隆区带等。
1.6 分组 根据患儿的血清型,将患儿分为AQP4-IgG阳性组、MOG-IgG阳性组和血清学阴性组。诊断不明确的中枢神经系统脱髓鞘患儿同时检测AQP4-IgG、MOG-IgG 2种抗体,当1种抗体阳性时,复发时再次检测这一种抗体;当2种抗体均阴性,每次复发时2种抗体均检测。起病至末次随访抗体检测均阴性的患儿为血清学阴性组,病程中只要有1种抗体阳性就归为检测阳性组。既往文献报道未见2种抗体同时阳性的情况。
1.7 统计学分析 使用SPSS 26.0进行数据录入和统计学分析。起病年龄、末次随访年龄、病程等计量资料用中位数(最小值,最大值)表示。比较不同血清型NMOSD随访时间、发作次数、末次随访EDSS评分有无差异时使用Kruskal-WallH检验,比较不同血清型NMOSD临床表型、影像学受累部位有无差异时使用χ2检验或Fisher精确检验。P<0.05为差异有统计学意义。
2 结果
2.1 一般情况 符合本文纳入标准的NMOSD患儿46例,男17例,女29例,男女比例为1∶1.7,起病年龄为6.4(0.9,14.4)岁,起病至末次随访中位病程为4.5(0.6,13.0)年。表1显示,MOG-IgG阳性组21例,AQP4-IgG阳性组12例,血清学阴性组13例,3组起病年龄、起病至末次随访中位病程和发作次数差异均无统计学意义,3组性别构成比差异有统计学意义,AQP4-IgG阳性组以女孩多见。
2.2 首次发作临床表型 表1显示,3组ON、TM、延髓最后区症状、间脑症状、脑炎和其他症状差异均无统计学意义,大脑综合征和脑干症状差异均有统计学意义。大脑综合征在MOG-IgG阳性组(42.9%)和血清学阴性组(43.8%)常见,脑干症状在AQP4-IgG阳性组(33.3%)和血清学阴性组(23.1%)常见。
2.3 病程中所有发作的临床表型 表1显示,3组ON、间脑症状、脑炎和其他症状差异均无统计学意义,TM、大脑综合征、脑干症状和延髓最后区症状差异均有统计学意义。TM在AQP4-IgG阳性组占比最高(43.9%),大脑综合征在MOG-IgG阳性组(36.2%)和血清学阴性组(34.4%)常见,脑干症状在AQP4-IgG阳性组(17. 1%)和血清学阴性组(18.0%)占比较高,延髓最后区症状在AQP4-IgG阳性组(12.2%)常见。
2.4 头颅MR受累部位 表1显示,3组皮层、内囊、导水管/四脑室周围和小脑受累差异均无统计学意义,皮层下白质、脑室旁白质、胼胝体、丘脑、基底节和脑干受累差异均有统计学意义,皮层下白质受累在MOG-IgG阳性组和血清学阴性组占比较高,脑室旁白质受累在AQP4-IgG阳性组和血清学阴性组占比较高,胼胝体受累在AQP4-IgG阳性组中常见,丘脑受累在AQP4-IgG阳性组、MOG-IgG阳性组占比较大,基底节受累在MOG-IgG阳性组占比较大,脑干受累在AQP4-IgG阳性组占比较大。不同血清型NMOSD患儿头颅MR特点见图1。
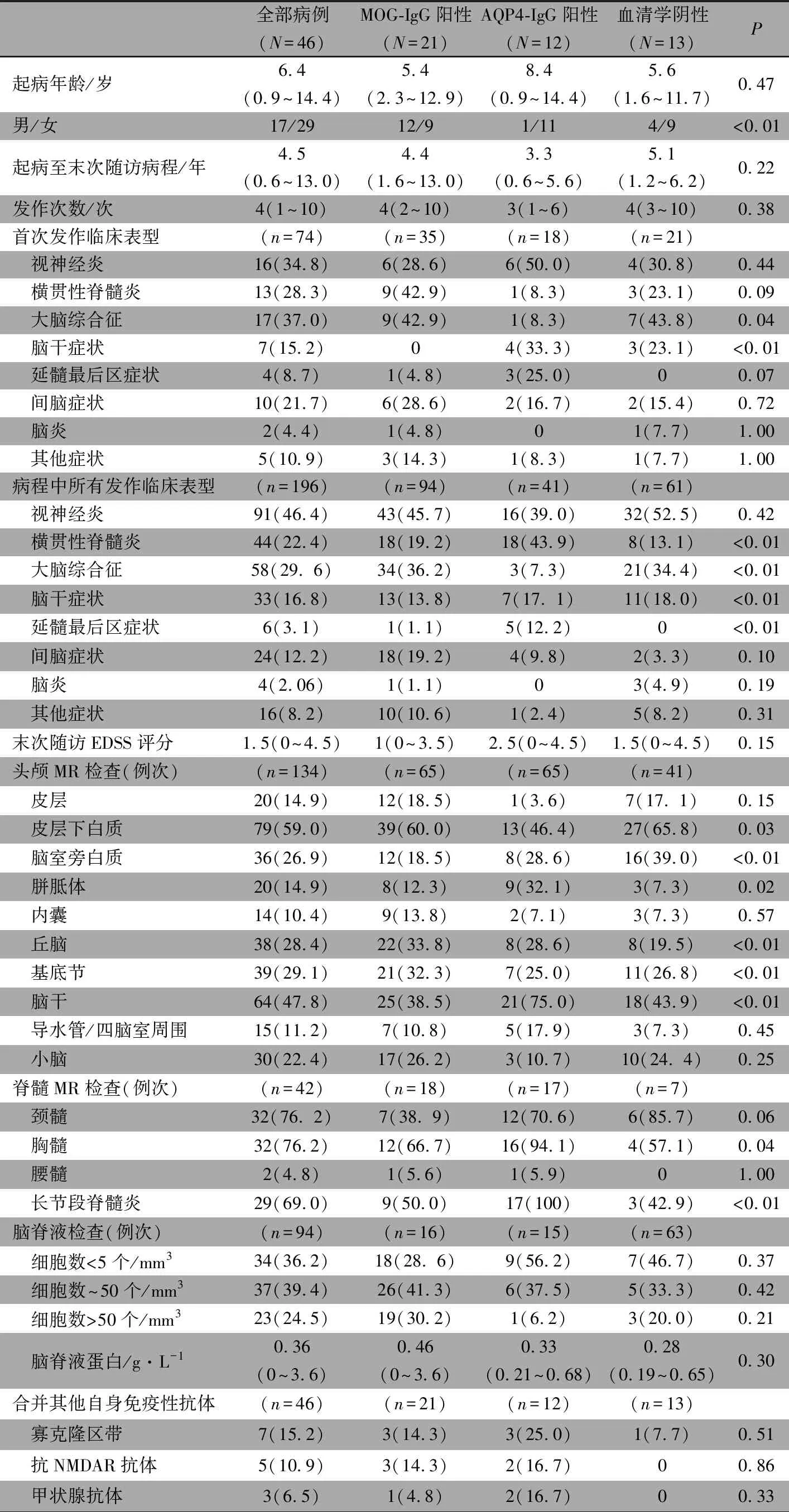
表1 不同血清型NMOSD临床表型、影像学和实验室检查的比较[n(%)]
2.5 脊髓MR受累部位 表1显示,3组颈髓和腰髓差异均无统计学意义,胸髓和长节段脊髓炎差异均有统计学意义,在3组中普遍受累,以AQP4-IgG阳性组更常见(94.1%和100%)。不同血清型NMOSD患儿脊髓MR特点见图1。
2.6 脑脊液检查情况 表1显示,共收集94例次急性期脑脊液, MOG-IgG阳性、AQP4-IgG阳性、血清学阴性患儿急性期脑脊液检查分别为16例次、15例次、63例次,46例次有核细胞数(个/mm3)<5 、~50、>50分别占36.2%、39.4%和24.5%,均以单个核细胞为主,蛋白中位数为0.36(0~3.6)g·L-1。3组脑脊液有核细胞数和蛋白差异均无统计学意义。
2.7 其他自身免疫性抗体检测情况 表1显示,共收集46例次自身免疫性抗体检测结果, 寡克隆区带阳性占15.2%、抗NMDAR抗体阳性占10.9%,甲状腺抗体阳性占6.5%。3组患儿不同自身免疫性抗体阳性比例差异均无统计学意义。
2.8 末次随访EDSS评分 46例均得到了末次随访EDSS评分,中位数为1.5(0~4.5)分,3组末次随访EDSS评分差异无统计学意义。11例MOG-IgG阳性患儿遗留神经系统后遗症,视觉能区8例,脑干能区2例,锥体能区、大脑能区和小脑能区各1例;9例AQP4-IgG阳性患儿遗留神经系统后遗症,视觉能区9例,锥体能区4例,直肠膀胱能区2例,感觉能区和大脑能区各1例;8例血清学阴性患儿遗留神经系统后遗症,视觉能区5例,锥体能区3例,大脑能区和直肠膀胱能区各1例。
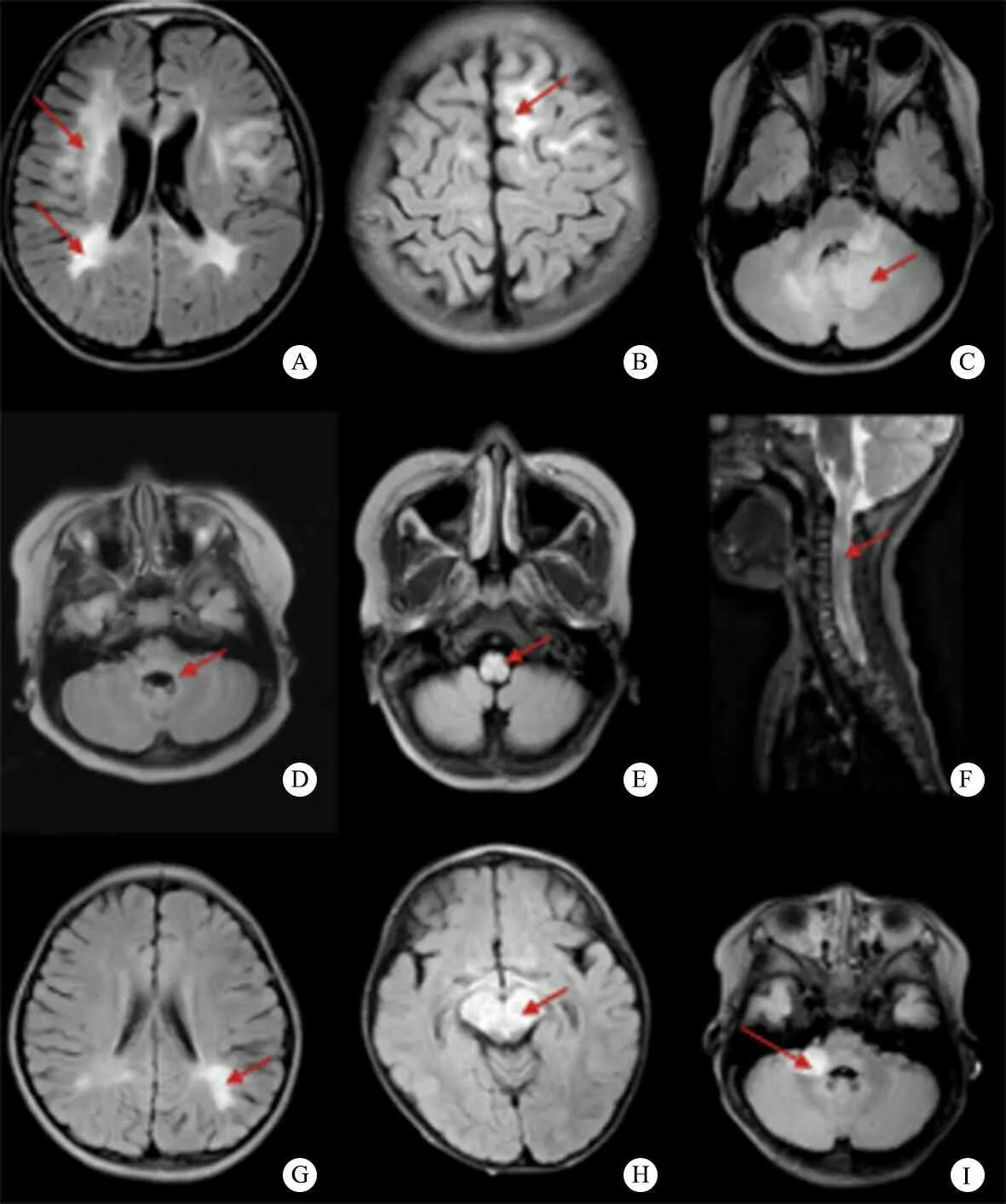
图1 不同血清型NMOSD患儿影像学特点
3 讨论
NMOSD的诊断主要根据核心症状并结合影像学,是一种临床-影像综合征。本研究共纳入46例NMOSD患儿,其中MOG-IgG阳性占45.6%, AQP4-IgG阳性占26.1%,2种抗体均阴性占28.3%。既往1项26例儿童NMOSD回顾性研究中,AQP4-IgG阳性为30%,MOG-IgG阳性为57%,血清学阴性为11%[5],与本研究相似。Alves等[6]回顾了多项成人NMOSD研究,AQP4-IgG阳性为70%~87%,MOG-IgG阳性为4%~11%,血清学阴性为14%~22%;AQP4-IgG阳性NMOSD男女比例为1∶10,MOG-IgG阳性及血清型阴性男女比例为1∶1。说明NMOSD患者中,儿童以MOG-IgG阳性多见,而成人以AQP4-IgG阳性更多见。与成人的队列研究类似,本研究中也证实AQP4-IgG阳性NMOSD以女孩居多,而血清学阴性和MOG-IgG阳性NMOSD无明显性别差异。
在首发表型和所有发作的临床表型中,ON常见于3种血清型NMOSD,TM在AQP4-IgG阳性组占比最高,既往文献报道MOG-IgG阳性NMOSD易累及视神经前段(视乳头和球后视神经)和脊髓下段(腰段和圆锥),更易同时出现视神经和脊髓受累[7,8]。本研究发现,大脑综合征常见于MOG-IgG阳性和血清学阴性患儿,表现为意识水平下降,认知、语言功能减退,头痛等,幕上病变为主,多呈ADEM样;延髓最后区症状见于12%的AQP4-IgG阳性儿童的临床发作,较少见于其他2种血清型儿童。既往儿童NMOSD研究中,大脑综合征症状见于67%的 MOG-IgG阳性病例,而仅见于16%~32%的 AQP4-IgG阳性患儿,延髓最后区症状在AQP4-IgG阳性NMOSD更具特异性,AQP4-IgG阳性患儿较MOG-IgG阳性患儿更容易出现孤立的脑干症状[9,10]。
在影像学特点中,本研究发现,皮质下白质受累在MOG-IgG阳性和血清学阴性患儿更常见,基底节受累在MOG-IgG阳性患儿更多见,脑干受累常见于AQP4-IgG阳性患儿。既往研究中AQP4-IgG阳性NMOSD的MR特征包括长节段脊髓炎(LETM)、下丘脑和脑室周围脑干病变,以及延髓背侧/最后区病变[1]。儿童髓鞘少突胶质细胞糖蛋白抗体相关疾病(MOGAD)的MR特征50%以上为皮层下白质受累,病灶较大,边界不清晰,丘脑和基底节受累多见[11,5]。既往报道小脑病变多见于MOG-IgG阳性患儿[5],本研究中小脑病变也多见于MOG-IgG阳性。颈段脊髓在3种血清型患儿均最常见,LETM在AQP4-IgG患儿中最常见。1项115例成人MOG-IgG阳性(40%,46/115)及AQP4-IgG阳性(60%,69/115)TM队列研究中,AQP4-IgG阳性TM表现为LETM较MOG-IgG阳性(86%vs52%)患者更多见,在随访中,脊髓纵向广泛的病变呈现出斑片状的外观或纵向广泛的脊髓萎缩[12]。本研究也表明,AQP4-IgG阳性以LETM更多见。约50%的MOG-IgG阳性患儿可有脊髓病变,大部分位于颈胸髓[11],MOG-IgG阳性患儿脊髓圆锥受累比AQP4-IgG阳性患儿(39%vs12%)更为常见[12]。
AQP4-IgG阳性患儿和MOG-IgG阳性患儿之所以存在上述表型差异,与MOG和
AQP4抗原的分布不同有关。MOG分布于中枢神经系统髓鞘表面,而AQP4主要分布在软脑膜、脑室周围、导水管周围、视神经以及脊髓中央灰质区,集中表达在血脑屏障周围,呈线性排列在星形胶质细胞足突上。研究发现,AQP4-IgG相关ON比MOG-IgG相关ON视力下降更显著,可能与AQP4在视网膜中高表达,而MOG在视网膜中不表达有关[13-16]。
本研究不足与局限性:①动态双向队列研究,回顾性部分信息可能存在回忆偏倚;②随访时点为起病后6个月,病程中所有发作的临床表型不一定能展现完全;③作为临床随访,要求6个月随访1次,难以保证复发或不复发病例的规律随访。
