VHL基因新突变致VHL综合征伴脑膜瘤一家系分析
2021-12-14王楚楚齐光照张丽侠王志芳许莉军郑丽丽秦贵军栗夏莲
李 冲,王楚楚,齐光照,张丽侠,王志芳,许莉军,郑丽丽,秦贵军,栗夏莲
1)郑州大学第一附属医院内分泌及代谢科 郑州450052 2)郑州大学生命科学学院 郑州450001 3)郑州大学第一附属医院药学部 郑州450052
von Hippel-Lindau(VHL)综合征是一种罕见的涉及多系统的常染色体显性遗传性肿瘤综合征(OMIM 193300),发病率为1/50 000~1/36 000。VHL基因(ID:7428,https://www.ncbi.nlm.nih.gov/gene/7428)的体系或胚系突变导致多个器官发展成为良性、恶性肿瘤和囊肿,包括中枢神经系统血管网状细胞瘤、视网膜血管网状细胞瘤(HGB)、肾癌(RCC)或肾囊肿、嗜铬细胞瘤(PHEO)、胰腺肿瘤或囊肿和生殖系统囊肿等病变[1-3]。VHL综合征在65岁人群中的外显率超过90%[4]。该病最常见的死亡原因是中枢神经系统的血管网状细胞瘤和转移性肾癌及其并发症。突变个体发生神经系统病变的风险显著增加,常见幕下(小脑、脊髓和脑干)神经母细胞瘤[5],出现在幕上的病变较为罕见。本研究对1例VHL综合征合并右额叶脑膜瘤患者进行VHL基因变异家系分析,通过错义突变蛋白功能预测软件Polyphen-2和3D图对该突变进行功能预测,结合文献复习,探讨脑膜瘤和VHL综合征的关系。
1 对象与方法
1.1 病例资料患者,男,30岁,以“发作性心悸、头痛、视物模糊19 a余,视物模糊再发3个月”为主诉于2020年3月收住郑州大学第一附属医院内分泌及代谢科。19 a前(2001年)无诱因出现发作性心悸、头痛、恶心,伴视物模糊,至当地医院测血压200/100 mmHg(1 mmHg=0.133 kPa)。眼底出血,肾上腺皮髓质功能未查,彩超示右肾上腺区实性占位(5 cm×2.6 cm×3.5 cm),行“右侧肾上腺嗜铬细胞瘤摘除术”。之后监测血压均正常。15 a前(2005年)在当地医院查腹部彩超和CT显示左侧肾上腺区实性占位,再次行左侧肾上腺嗜铬细胞瘤摘除术”。4 a前(2016年)在当地医院查腹部彩超、增强CT和SPECT显示右侧肾上腺区、腔静脉前门脉后嗜铬细胞瘤可能,行“腹腔镜下异位嗜铬细胞瘤切除术”,病理诊断为(副主动脉旁)副节瘤。3个月前(2019年12月)再次出现左眼视物模糊,当地诊断为左眼底出血,腹部CT显示腹膜后嗜铬细胞瘤术后改变,左侧肾上腺区多发病灶,考虑嗜铬细胞瘤可能。自发病以来,神志清,精神可,大小便正常,体重无明显变化。既往史、个人史、婚育史无特殊。体格检查:除手术瘢痕外,未见异常。
1.2 家系概况及VHL基因检测收集患者(先证者)外周血及病理组织切片。患者自述非近亲结婚。父亲及奶奶血压高,服药效果好。患者有1兄1弟1姐及2女,均体健,测血压均正常。获得患者及其家属同意后收集5人的外周血样本,进行VHL基因检测。
1.2.1基因组DNA的提取 采用康为世纪生物科技有限公司的血液基因组DNA Mini试剂盒提取外周血DNA。采用南京诺唯赞生物科技有限公司的FastPure®FFPE DNA Isolation试剂盒提取病理组织切片基因组DNA。提取过程中的配套器材及操作流程严格按照说明书进行。
1.2.2VHL基因编码区的扩增 用Primer 5.0软件设计3对引物,采用南京诺唯赞生物科技有限公司提供的2×Taq Plus Master Mix Ⅱ(Dye Plus)试剂盒进行PCR扩增;引物序列见表1。PCR反应体系:2×Taq Plus Master Mix Ⅱ(Dye Plus)12.5 μL,上下游引物(10 μmol/L)各1 μL,基因组DNA模板(15~25 mg/L)1 μL,ddH2O补至25 μL。

表1 PCR引物序列
1.2.3PCR产物测序 PCR产物经琼脂糖凝胶电泳后进行目的条带纯化,然后在郑州普利莱医学检验所实验室进行测序。应用Sanger测序技术对患者外周血及病理组织切片进行VHL基因全部编码区测序。使用Chromas及Sequencing Analysis Version 5.0对测序结果进行分析,测序结果与GenBank数据库中序列比对。
1.2.4蛋白功能预测 利用软件Polyphen-2(www.genetics.bwh.harvard.edu/pph2)对错义突变蛋白进行功能预测。
2 结果
2.1 实验室、影像学和病理学检查结果患者的血尿粪常规、肝肾功、电解质、甲状腺功能、促甲状腺激素(TSH)、肾上腺髓质功能检查均未见异常。全天血压动态变化呈弱杓型曲线。眼底检查见图1A,全腹部CT平扫+增强检查结果见图1B,肾上腺髓质SPECT/CT融合显像结果见图1C,头颅MRI平扫与增强结果见图1D。患者于2020年3月在我院神经外科行“右额叶肿块切除术”。术后病理显示(右额叶占位)脑膜瘤,WHOⅠ级。免疫组化结果:CK(-),S-100(-),EMA(-),SSTR2(+),PR(部分+),GFAP(-),Ki-67(2%+),CD34(血管+),ERG(血管+),NSE(-),Inhibi-a(-),STAT6(-),CgA(-),Syn(-),D2-40(血管+)。

A:眼底照相显示左眼视盘表面可见橘红色圆形隆起病灶,左眼眼底荧光血管造影提示视网膜毛细血管瘤;B:肾上腺CT显示左侧肾上腺区多发病灶;C:肾上腺髓质SPECT/CT融合显像,静脉注射显像剂131I-MIBG后,左侧肾上腺可见软组织结节影,部分放射性分布浓聚,大者约1.3 cm×1.5 cm,CT值约41 Hu;腹膜后可见软组织结节影放射性分布浓聚,大小约1.4 cm×1.9 cm,CT值约27 Hu;D:头颅MRI显示右侧额叶见类圆形长T1长T2信号,内见多发小圆形长T1长T2信号,T2 Flair序列部分呈高信号,高B值DWI可见点片状扩散受限高信号,病变大小约26 mm×26 mm×21 mm(前后径×左右径×上下径)图1 患者影像学资料
2.2 VHL基因检测结果及分析患者血液和病理组织标本的测序结果均提示患者携带1个VHL基因的杂合错义变异(c.284C>G),该变异导致第95位编码氨基酸残基由脯氨酸Pro变为精氨酸Arg(p.Pro 95 Arg)。患者父母及一个女儿均未携带该变异,提示该变异为新生变异。另一个女儿为该变异杂合子携带者,其测序结果见图2A。
c.284C>G变异未被ClinVar数据库收录,但收录在HGMD数据库中,并在1例嗜铬细胞瘤患者中有过报道[6]。该变异附近氨基酸残基的改变(p.Asn 90 Ile;p.Gly 93 Cys;p.Gly 93 Arg;p.Gly 93 Asp;p.Tyr 98 His;p.Tyr 98 Cys)与VHL综合征的致病或可能致病均已有报道[7-9],表明该变异位于热点突变区。此外,该变异频率在ExAC、GenomAD、1000G参考数据库中未见收录,不属于多态性变化。c.284C>G编码蛋白p.Pro 95 Arg所在区域片段的物种保守性分析显示p.Pro 95在智人、恒河猕猴、老鼠、狗、非洲大象和鸡这些物种中高度保守(图2B),位于VHL第一外显子(图3A)。经过错义突变蛋白功能预测软件Polyphen-2预测,该突变对VHL蛋白造成显著影响,预测的氨基酸突变为probably damaging,评分为0.994(图3B)。在蛋白质3D图上,突变位点c.284C>G位于VHL的功能区,与缺氧诱导因子(HIF)结合,具有非常重要的功能(图3C)。结合在家系验证中其父母未携带该变异的情况,根据ACMG指南判定该变异为可能致病,会对基因或基因产物造成有害的影响。
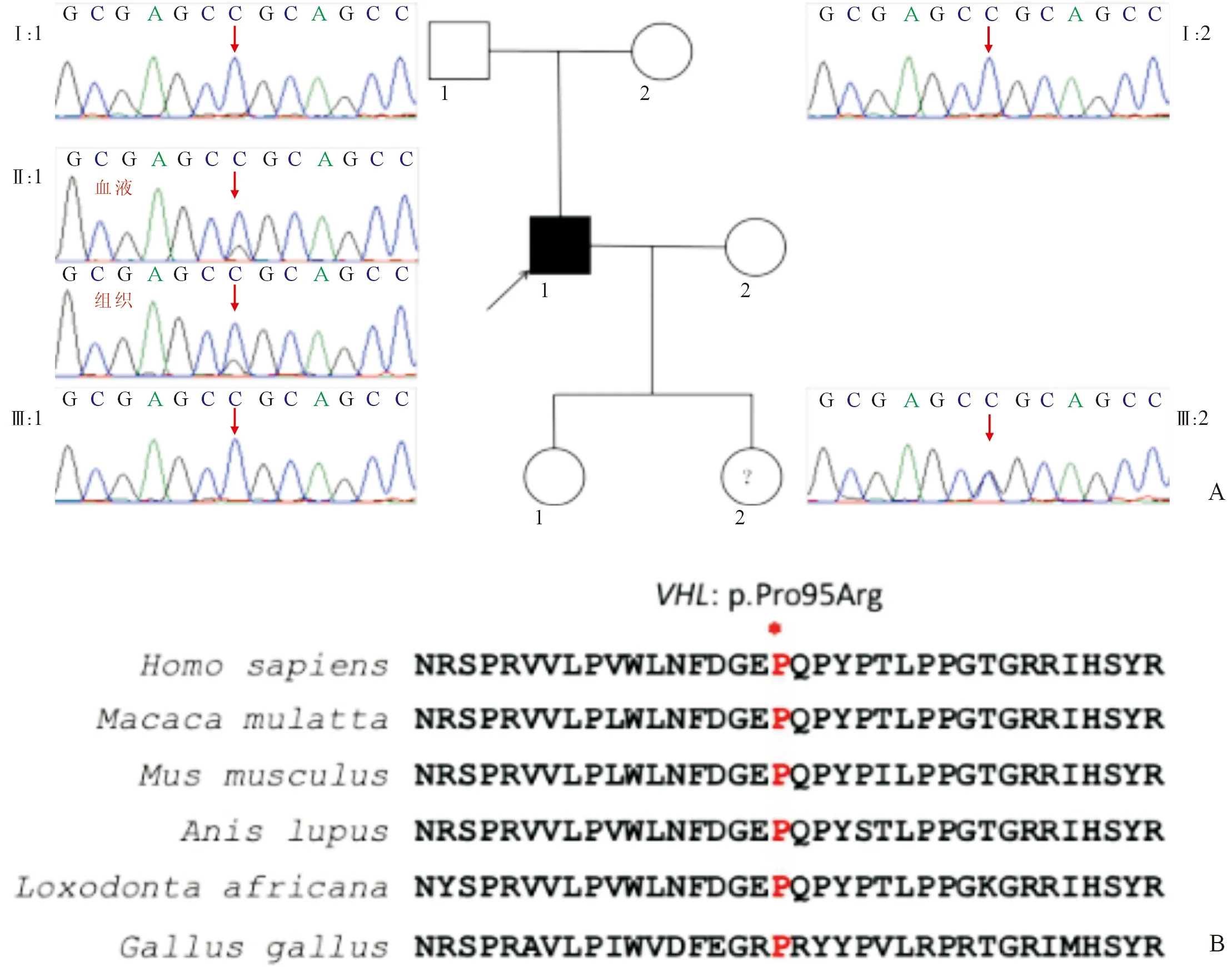
A:红色箭头示变异位置,先证者(Ⅱ:1)病理样本及血液样本测序结果均提示携带变异(c.284C>G);父(Ⅰ:1)、母(Ⅰ:2)、女儿1(Ⅲ:1)未携带该变异;女儿2(Ⅲ:2)携带该变异;B:VHL:p.Pro 95 Arg所在编码蛋白质区域片段的物种保守性分析显示p.Pro 95在物种间高度保守图2 先证者VHL基因测序图
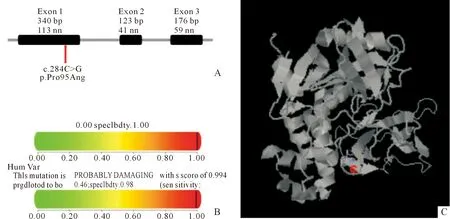
A:c.284C>G位于VHL第一外显子;B:Polyphen-2预测显示该突变对VHL蛋白造成显著影响,预测的氨基酸突变为probably damaging,评分为0.994;C:在3D图上,其蛋白产物位于VHL的功能区,与HIF结合,具有非常重要的功能图3 VHL基因 c.284C>G突变的蛋白功能预测
3 讨论
VHL是一种重要的肿瘤抑制因子,参与HIF的泛素化和降解,在氧调控基因表达中起着重要作用。VHL位于染色体3p25.3,包括3个外显子,编码蛋白是蛋白质复合物的组成部分,包括延伸蛋白B、延伸蛋白C、Cullin-2和具有泛素连接酶活性的E3。VHL突变导致蛋白失活,从而使下游底物上调,促进一系列致癌基因的表达,是多器官肿瘤发生的重要机制[10]。
VHL综合征具有典型的基因型-表型相关性。根据是否合并嗜铬细胞瘤,分为两型:1型无嗜铬细胞瘤,常由VHL外显子缺失、截断、移码或无义突变引起;结合是否合并肾癌,分为1A型(合并肾癌)和1B型(无肾癌)。2型包含嗜铬细胞瘤,多由VHL错义突变引起;进一步又可分为肾癌风险低的2A型、肾癌风险高的2B型和只有嗜铬细胞瘤表现的2C型[1,11]。
本文报道的VHL综合征患者11岁因高血压陆续发现左侧肾上腺和腹膜后嗜铬细胞瘤,此次因左眼视物模糊入院,完善FFA(眼底荧光血管造影)后发现左眼视网膜毛细血管瘤,综合考虑可能为VHL综合征,进一步对血液和嗜铬细胞瘤病理组织进行基因检测,显示该患者携带VHL杂合错义突变c.284C>G,导致第95位氨基酸由脯氨酸Pro变为精氨酸Arg(p.Pro 95 Arg),为胚系突变。经不同物种氨基酸序列比对,第95位脯氨酸在多个物种中具有高度保守性。VHL功能缺失等同于细胞缺氧状态,这可能是导致该患者发生多种肿瘤的分子机制。
进一步检查发现右侧大脑额叶占位,术后病理显示脑膜瘤。经过文献检索,脑膜瘤与NF2杂合缺失有关,在VHL综合征中非常罕见[12],近年来仅有少数VHL综合征合并脑膜瘤的病例报道,Kanno等[13]报道的一例VHL综合征2B型合并脑膜瘤患者在VHL第三外显子685位胞嘧啶突变为鸟嘌呤(685C>G)导致158位亮氨酸突变为缬氨酸(L158V)(胚系突变)。Santarpia等[14]描述一例26岁VHL综合征2C型患者,先后经历了双侧肾上腺嗜铬细胞瘤、腹主动脉和膀胱副神经节瘤切除术,后来发现右侧额叶脑膜瘤,基因检测显示VHL695G>A导致161位精氨酸突变为谷氨酰胺(R161Q)。在本研究中,该患者为VHL综合征2C型,存在VHL第一外显子的杂合错义突变(c.284C>G),通过Polyphen-2和蛋白结构3D图对错义突变c.284C>G的功能进行预测,提示该突变是功能突变,能显著影响VHL的活性,与临床表型关系密切,但是否导致其罹患右侧额叶脑膜瘤尚不清楚。
随后我们对患者的家系成员进行突变位点的检测,患者一8岁女儿携带VHL c.284C>G,目前并未发现有相关肿瘤及囊肿的发生。根据国内外指南,对于无症状VHL基因突变携带者目前尚无有效的干预肿瘤发生的手段,只能进行密切监测[15]。由于视网膜肿瘤的发生风险是在青少年时期,我们正在对该女进行定期随访、预防性监测,待成年后进行遗传咨询,以期早期发现肿瘤,及早治疗,提高其生活质量和预期寿命。
VHL突变的多样性,相同突变位点外显率和发病年龄的显著差异给VHL综合征的诊治带来了巨大的挑战[16]。Iida等[17]对存在VHL基因695G>A(R161Q)突变的VHL综合征2A型先证者的16位家庭成员进行检测,发现8例携带相同突变位点的基因,其中一例表现为胰腺神经内分泌肿瘤,无嗜铬细胞瘤,同卵双胞胎中的一例表现为巨大的嗜铬细胞瘤和视网膜母细胞瘤,而另一例则无任何表型。目前尚不能通过基因检测来预测患者可能出现的表型,比如哪个器官会受累,发生风险的高低,肿瘤在特定生命阶段(幼儿期、妊娠期等)的特殊发生,或肿瘤生长的速度等。VHL基因的胚系突变与产生的临床表型之间的相关性取决于突变的VHL下调HIF途径活性的能力,此外,年龄、性别、基因型、妊娠、肿瘤的位置等都可能影响患者的临床表型[18]。近年来,许多与HIF无关的VHL功能被发现,这进一步提示了VHL综合征表型调控机制的复杂性。
总之,我们发现了VHL综合征中一个新的胚系突变VHL c.284C>G,对VHL的功能可能造成显著影响,该突变与脑膜瘤发生发展的相关性是我们未来进一步研究的焦点。同时,本课题组正在开展对无症状VHL突变携带人群进行密切随访和实施潜在干预的临床研究,以提高VHL综合征患者的生活质量,改善其预后。
