SPTB基因杂合突变致遗传性球形红细胞增多症1例
2021-12-14张志华张荣娟郝长来
温 雪,张志华,张荣娟,郝长来
(承德医学院附属医院 血液内科,河北 承德 067000)
遗传性球形红细胞增多症(HS)是一种具有明显异质性的家族性溶血性疾病。HS作为一种常见的遗传性溶血性疾病,是北欧和北美最常见的遗传性慢性溶血的原因,据报道发病率为1/2000[1],我国人群发病率约1/10万[2]。本病临床症状多种多样,从无症状贫血到溶血性危象,妊娠、突然失血或叠加感染均可加重。但因其症状不典型, 如没有症状的携带者临床没有溶血征象,但红细胞渗透脆性增加,有遗传性球形红细胞的基因突变,后代可以发生HS;或轻型的HS患者由于其骨髓代偿功能好,可以没有或仅有轻度贫血和脾肿大,血清胆红素和网织红细胞计数轻度增高,外周血球形红细胞比较少见。故实验室检查易受影响, 误诊、漏诊率高[3]。本研究就1例临床上表现为全血细胞减少的患者,应用高通量测序技术以及Sanger测序法确诊为HS 2型患者进行报告如下。
1 临床资料
患者,女,29岁,主因发现贫血5年,发热、乏力2天于2020年3月16日就诊我院。既往5年前因“妊高症”行剖宫产术,有输血史。家族中其他成员无类似病史。入院查体:患者神清语利,贫血貌。皮肤、黏膜苍白,浅表淋巴结未触及肿大,巩膜无黄染,肝脾触诊不满意,双下肢无水肿。辅助检查:血常规+网织细胞计数:白细胞计数1.28×109/L,红细胞计数1.19×1012/L,血红蛋白44 g/L,血小板计数48×109/L,网织红细胞百分比13.8%,平均红细胞体积(MCV)118.6 fL,平均血红蛋白含量(MCH)39.7 pg,平均血红蛋白浓度(MCHC)335 g/L,叶酸17.16 nmol/L,维生素B12 164.2 pmol/L, 血涂片检查:中性分叶核粒细胞90%,淋巴细胞9%,单核细胞1%。溶血相关检查:蔗糖溶血试验、血清酸化溶血试验(Ham)、直接抗人球蛋白试验(Coombs)、红细胞渗透脆性试验、冷凝集试验,均未见异常,PNH免疫分型:红细胞CD59缺失百分率0.83%;粒细胞中FLAER缺失细胞百分率0.00%。腹部CT:脾大。骨髓象(髂骨):增生活跃(-),原始细胞未见,红系增生减低,巨核细胞不少。骨髓免疫分型:大致正常。骨髓病理活检:HE及PAS染色示骨髓增生较低下(50%),粒红比例大致正常,粒系各阶段细胞可见,以中幼及以下阶段为主,红系各阶段细胞可见,以中晚幼红细胞为主,巨核细胞数量大致正常;少量淋巴细胞散在分布。网状纤维染色(MF-1级)。诊断全血细胞减少症,给予叶酸、腺苷钴胺等药物治疗,监测血常规+网织细胞计数:白细胞计数5.26×109/L,血红蛋白77 g/L,血小板计数94×109/L,网织红细胞百分比14.0%,MCV 114.3 fL,MCH 37.8 pg,MCHC 331 g/L,叶酸34.28 nmol/L,维生素B12 484 pmol/L。肝功能:总胆红素27.9 μmol/L,非结合胆红素27.2 μmol/L,血清总胆汁酸15 μmol/L。复查骨髓象:增生明显活跃,粒系比例减低,红系明显增生,可见花瓣核、畸形核幼红细胞,成熟红细胞大小不一。共见巨核细胞36个。髓系肿瘤二代测序:阴性,诊断为骨髓增生异常综合征(MDS)?给予环孢素治疗后,监测血常规+网织细胞计数:白细胞计数4.97×109/L,红细胞计数1.50×1012/L,血红蛋白59 g/L,血小板计数74×109/L,网织红细胞百分比16.1%, MCV 103.4 fL, MCH 33.3 pg, MCHC 322 g/L,肝功能:总胆红素72.06 μmol/L,直接胆红素19.20 μmol/L,因其治疗过程中感染好转,白细胞降低至正常值,血小板升高但仍低于正常值,贫血依旧,考虑为溶血性贫血,建议患者行相关基因检测。见图1。
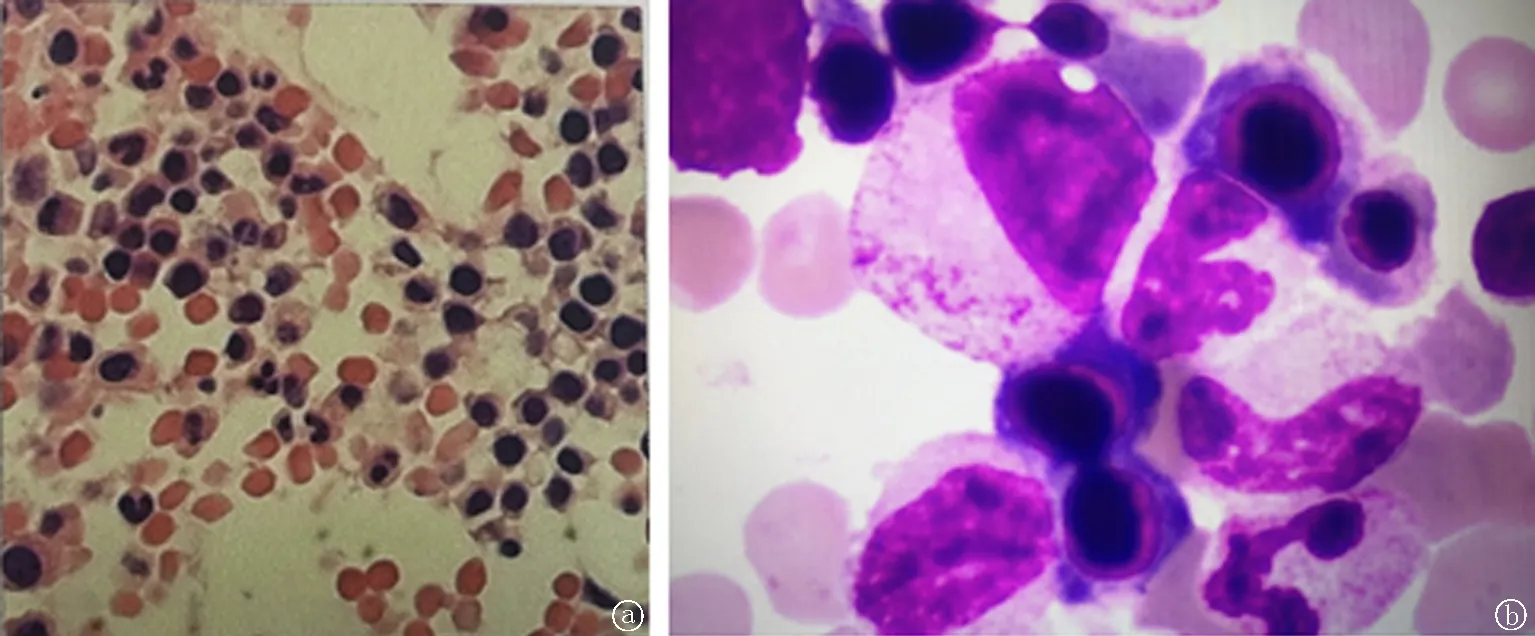
图1 患者骨髓形态学检查 a.骨髓病理活检; b.骨髓象分析
基因检查:采集患者及其父亲、母亲外周血标本4 ml(EDTA抗凝),对患者进行高通量测序,其父亲、母亲进行Sanger验证。经检测发现患者SPTB基因有1个杂合突变: 在5551号核苷酸由胞嘧啶 C变为胸腺嘧啶 T(c.5551C>T)的杂合突变,导致氨基酸发生无义突变(p.Q1851X)。经家系验证分析,c.5551C>T(p.Q1851X)受检人之父该位点无变异,受检人之母该位点无变异,故此变异为自发突变。美国医学遗传学与基因组学学会(ACMG)遗传变异信息详细解读:c.5551C>T(exon26,NM_001355436),导致氨基酸改变p.Q1851X,该变异为零效变异(无义突变),可能导致基因功能丧失。根据 ACMG指南,该变异初步判定为 致病性变异(pathogenic),且在正常人群数据库中的频率为(-),为低频变异;文献数据库未有该位点的相关性报道 ClinVar 数据库无该位点致病性分析结果。患者同时合并存在ANK1基因杂合突变c.1495G>T(p.A499S),受检人之父该位点杂合变异,受检人之母该位点无变异,但其父亲无相关临床症状,根据 ACMG指南,该变异初步判定为临床意义未明,故此为非致病基因。见图2~3。

图2 患者及其父母SPTB基因测序 a.患者SPTB 基因杂合突变 c.5551C>T(p.Q1851X);b、c.患者父亲、母亲 SPTB 基因型为野生型
2 突变基因的蛋白质结构分析
使用SWISS-MODEL软件(http://swissmodel.expasy.org)对SPTB基因突变进行同源建模。使用模型为3kbu.1.A,序列同源性为99.07%,相似性为61%。蛋白结构预测发现c.5551C>T(p.Q1851X) 位点变异导致蛋白截断突变。截断突变是那些使蛋白质产物变短因而弱化或破坏蛋白质功能的某些突变,这种截断突变包括无义突变和移码突变。研究发现移码突变是膜结合域和膜收缩蛋白结合域中最常见的突变类型,将导致成熟前翻译终止信号的出现;而无义突变则是调节域中最常见的,可致使基因编码的蛋白质失去原有的功能[4]。见图4。
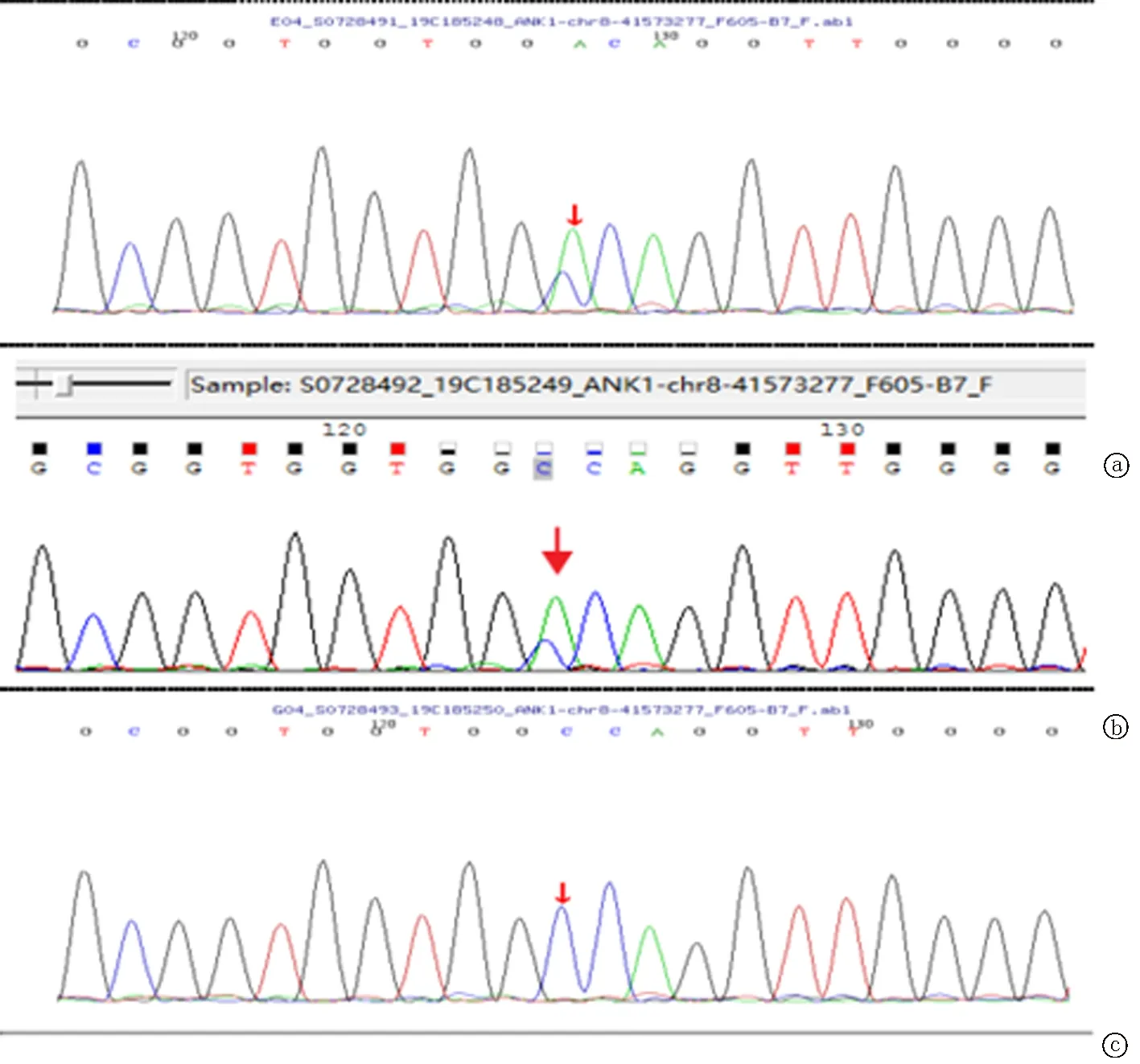
图3 患者及其父母ANK1基因测序 a.患者ANK1基因杂合突变c.1495G>T(p.A499S);b.患者父亲ANK1基因在该位点为杂合变异(无致病性);c.患儿母亲ANK1 基因型为野生型

图4 SPTB基因突变前后的蛋白三维结构预测 a.野生型结构; b.突变型结构
3 讨 论
HS是一种常见的遗传性溶血性贫血,因外周血中出现球形红细胞而得名。其特点为血液中包含大量球形红细胞,临床症状的严重程度、膜蛋白缺陷和遗传模式存在广泛的异质性[5]。在所有HS患者中,约75%为常染色体显性遗传,15%为常染色体隐形遗传,无家族史的散发病例可能为新发生的基因突变所致[6]。病理基础为红细胞膜蛋白基因异常,致膜骨架蛋白缺陷,细胞膜脂质丢失,细胞表面积减少,细胞球形变,通过脾脏时容易被破坏。目前脾切除术是治疗HS的唯一方法。
HS的发病机制为基因缺陷导致的红细胞膜蛋白异常[7],与HS红细胞膜和脂质双分子层相互作用相关的基因有5个,包括:SPTA1、SPTB、ANK1、SLC4A1和EPB42,分别导致锚蛋白、β收缩蛋白、带3蛋白、4.2蛋白和α收缩蛋白缺陷或异常[8]。≥1个HS相关基因突变可导致膜蛋白缺失,进而导致HS[9]。这5个基因的突变在国家间存在差异,欧美人群最常见的是ANK1 基因突变,约占 HS 半数。我国HS 患者常见的突变基因为ANK1 和SPTB, 主要表现为错义突变和无义突变。HS 相关基因突变与严重程度之间无显著相关性[10],因此HS在临床上容易被误诊和忽略。
HS典型的临床表现有贫血、溶血性黄疸、脾大,感染可使病情加重,常伴胆石症、痛风、顽固的踝部溃烂或下肢红斑性溃疡等并发症。典型的血常规为血红蛋白和红细胞轻度或中度降低,发生危象时可呈重度;白细胞和血小板正常;网织红细胞升高;MCV和MCH多正常,MCHC可增加[11-12]。该患者入院时发热、乏力,血红蛋白重度减少,考虑感染引起的溶血危象导致全血细胞减少。给予对症处理,治疗过程中感染好转,白细胞降低至正常值,血小板升高但仍低于正常值,贫血依旧,考虑溶血性贫血。因其血小板低于正常值,红细胞渗透脆性试验正常,均与溶血性贫血不符,故外送患者外周血标本进行基因检测。
该病例全血细胞减少,临床表现不典型,常规实验室检查均无法明确诊断,因此进一步完善基因检测对疾病精准诊断具有重要意义。初诊时表现为全血细胞减少,为大细胞性贫血,叶酸和维生素B12为正常低值,考虑到有可能是营养不良贫血,试验性地补充叶酸和维生素B12后, 血象有明显改善,可知患者有营养不良因素存在,且存在病态造血,感染体征,脾脏肿大等,故考虑不除外MDS。后加用环孢素治疗后感染好转但依旧贫血,且根据基因检测结果可知,该患者为SPTB基因的致病性变异,不符合MDS的诊断标准,依据HS的发病机制,尤其在溶血时可以出现叶酸和维生素B12缺乏,可考虑为不典型HS。根据SWISS-MODEL软件对SPTB基因突变进行蛋白结构预测可知,c.5551C>T(p.Q1851X) 位点变异导致蛋白截断突变,SPTB基因突变可能使β-血影蛋白缺乏,破坏了红细胞膜的稳定性,导致红细胞破坏, 也可说明该患者为HS。因HS中无血小板减少,所以需进一步随诊监测,随诊:患者行脾脏切除术,术后3个月复查血常规均正常。
综上所述,HS患者的临床表现无特异性,部分临床表现不典型发作的HS诊断具一定挑战性,排除其他溶血性贫血及其他相关因素后,建议考虑HS。此外,本研究发现SPTB基因的1个未见报道的突变,丰富了该基因突变数据库,为进一步探索HS的遗传学病因提供参考。
