14 种帘蛤科贝类线粒体基因组特征与系统进化分析
2021-12-13颜成瑞叶莹莹
颜成瑞,苗 菁,叶莹莹
(国家海洋设施养殖工程技术研究中心,浙江舟山 316022)
帘蛤科Veneridae 隶属于软体动物门,双壳纲,帘蛤目,在世界分布面较广,且数量大,经济种类多,目前已知有500 多种,我国常见的有60 余种,常见物种如文蛤Meretrix meretrix (Linnaeus,1758),菲律宾蛤仔Ruditapes philipinarum,等边浅蛤Macridiscus Multifarius 等作为我国贝类养殖业的主要成员,具有较高的经济价值[1-2]。然而关于帘蛤科贝类系统进化的研究最初基于形态以及古生物学证据,随着现代分子技术的逐渐发展,学界认为帘蛤科贝类的形态趋同进化对系统关系的研究存在严重误导,现行的帘蛤科贝类的分类系统无法代表真实有效的进化关系。对于帘蛤科贝类在生活习性,形态上相似等问题,近年来众多学者通过分子生物学手段,如DNA 分子标记等方法来对帘蛤科贝类进行遗传多样性,系统发育及生物资源保护等方面的研究[3]。如程汉良等[4]以16S rRNA 序列进行分析,对中国沿海21 种帘蛤科贝类进行研究,阐明了帘蛤科分类和系统演化的一系列问题,为帘蛤科分类提供证据;陈军[5]选取135 种双壳贝类,其中帘蛤科种类128 种,帘蛤超科5 种,其他双壳贝类外群2 种,通过2 个线粒体基因序列COI,16S rRNA 和一个核蛋白编码基因H3 进行分析,对帘蛤科内中国沿海某些类群进行了修订;田云方[6]通过COI 和Cytb 序列,对我国沿海帘蛤科中的斧文蛤,文蛤,等边浅蛤群体进行分析,表明了不同海域帘蛤科群体的遗传差异较大;陈道海等[7]利用COI 序列对沿海10 属10 种帘蛤科贝类进行分析,证明了COI 序列适用于帘蛤科贝类种群的分子系统发生学研究。而线粒体基因伴随着核基因组DNA 共同进化,同时也记录了丰富的历史信息。通过对线粒体基因组DNA 的分析比较,可以了解到线粒体DNA 不同基因在进化过程受到选择压力的大小。因此线粒体DNA 是研究物种遗传多样性与进化的有力工具之一[8]。
鉴于目前尚未有基于线粒体全基因组分析帘蛤科系统发育的研究,本研究运用线粒体基因组全序列对帘蛤科贝类进行分析,选取NCBI 上所能下载到的帘蛤科5 属14 种贝类,对其线粒体基因组基本特征,蛋白质编码基因,基因排序,选择压力进行分析,并建立系统发育树,以期为帘蛤科贝类亲缘关系,系统发育,种质资源等研究提供遗传信息。
1 材料和方法
1.1 数据获取
从Genbank 数据库查询下载14 种帘蛤科贝类线粒体DNA 全序列,这14 个物种分别是文蛤属Meretrix 的文蛤M.meretrix、斧文蛤M.lamarckii、丽文蛤M.lusoria、皱肋文蛤M.lyrata、中华文蛤M.petechialis;镜蛤属Dosinia 的高镜蛤Dosinia altior、日本镜蛤D.japonila、射带镜蛤D.troschheli;巴非蛤属Paphia 的和蔼巴非蛤Paphia amabilis、真曲巴非蛤P.euglypta、织锦巴非蛤P.textile、波纹巴非蛤P.undulata;石房蛤属Saxidomus 的紫石房蛤Saxidomu spurpuratus,浅蛤属Macridiscus 的等边浅蛤Macridiscus multifarius。
1.2 数据处理
采用BioEdit 软件对全部序列进行比对编辑,得到14 个物种线粒体基因组全长,并通过DAMBE 软件统计序列碱基组成含量及比例;同时列出14 个物种13 个蛋白质编码基因的起始密码子,终止密码子和氨基酸数量。同时将14 个物种的蛋白质编码基因以COI 为首进行排列来研究基因重排;利用MEGA 6[9]软件基于12 个蛋白质编码基因串联后的序列计算不同物种之间的遗传距离;通过Ka/Ks-calculator 2.0 来计算12 个蛋白质编码基因的同义替换率和非同义替换率;并基于MEGA 6 中的J.C 参数模型(Jukes-cantor Model)构建邻接系统进化树,1 000 次重抽样检验系统树中节点的自引导值。
2 结果
2.1 线粒体DNA 序列特征及分析
14 种帘蛤科贝类线粒体DNA 全长在17 229 bp~21 625 bp 之间,其中射带镜蛤线粒体基因组长度最短,皱肋文蛤线粒体基因组最长。计算14 种帘蛤科贝类线粒体全基因组A,G,T,C 碱基含量,如表1 所示。A,G,T,C 碱基平均含量依次对应分别为26.58%,23.01%,40.67%,10.09%,14 种帘蛤科贝类均表现出C碱基含量最低,T 碱基含量最高,A 碱基较高于G 碱基含量的分布特征。所有序列A+T 碱基含量略高于G+C 碱基含量,表现出明显的AT 偏向[10-11]。

表1 14 种帘蛤科贝类线粒体基因组特征Tab.1 Basic characteristics of 14 species mitochondrial genomes of Veneridae
2.2 蛋白质编码基因特征分析
14 种帘蛤科贝类的12 个蛋白质编码基因中,有1 个ATP 合成酶亚基ATPase6,3 个细胞色素氧化酶的亚基COI,COII,COIII,Cytb,7 个NADH 脱氢酶亚基ND1-6 和ND4L(14 个物种中文蛤,中华文蛤,真曲巴非蛤,紫石房蛤,无ATP8 基因。采用DAMBE 软件分析其密码子组成情况(表2)。13 种基因编码氨基酸的数量在不同帘蛤科贝类中存在差异。其中同属的帘蛤科贝类大部分氨基酸数量相同或者具有微小差异,不同属的物种之间存在的氨基酸数量差异较大,ATG 并不是唯一的起始密码子[12]。文蛤线粒体基因组的COI、中华文蛤的COI、高镜蛤ND4L 和Cytb、射带镜蛤的COI 和COII,都是以TTG 作为起始密码子的;而斧文蛤的ND4L,ND5,Cytb、皱肋文蛤的ND1,ND2,ND4,ATP6,Cytb、中华文蛤的ND5,ND6,Cytb、高镜蛤的ND1,ND4、日本镜蛤的ND4L,ND5、射带镜蛤的ND1,ND4L,ND4,ND5、真曲巴非蛤的ND5,ND6、织锦巴非蛤的COI,Cytb、等边浅蛤的ND1,ND4,ND5,都是以ATA 作为起始密码子的;皱肋文蛤的ND3,COII,COIII、斧文蛤的ND6、镜蛤亚科的ND2,ND3、日本镜蛤的COI,COII、高镜蛤的ATP8、巴非蛤亚科的ND1,ND4L、真曲巴非蛤的ND3、COIII、波纹巴非蛤的ND5、和蔼巴非蛤的COIII 和Cytb,都是以GTG 作为起始密码子的;皱肋文蛤的ND5、高镜蛤的ND6,COI,COII、日本镜蛤的Cytb、射带镜蛤的Cytb、真曲巴非蛤的ND4、织锦巴非蛤和波纹巴非蛤的ND4,ND6,COII,COIII、和蔼巴非蛤的ND6、等边浅蛤的ND2,ND6,COI,都是以ATT 作为起始密码子的。而真曲巴非蛤的Cytb 是以GTT 作为起始密码子的。终止密码子有TAA 和TAG 两种。

表2 蛋白质编码基因的氨基酸数量和起始终止密码子Tab.2 The number of amino acids and the initation and termination codon of the protein-coding gene
2.3 基因重排
比较帘蛤科的14 个物种线粒体基因的排列,发现其中存在差异,表明了帘蛤科这14 个物种在进化过程中经历了一定规模的基因重排。由表3 可以看出,14 个物种在基因排列上共享5′-ND1-ND2-ND4LCOII-3′基因框。文蛤属内中华文蛤和文蛤,石房蛤属的紫石房蛤,巴非蛤属的真曲巴非蛤,与典型的后生动物线粒体基因组来说,缺失ATP8 基因。文蛤属,镜蛤属物种的基因排列顺序基本相同,和石房蛤属的紫石房蛤相比有较小差异,而与巴非蛤属相比的基因排列顺序差异较大。此外,值得注意的是浅蛤属的等边浅蛤的基因排列顺序与其他帘蛤科物种大不相同。由此也可以看出属与属之间亲缘关系的远近,且结论与系统发育树所展现的一致。
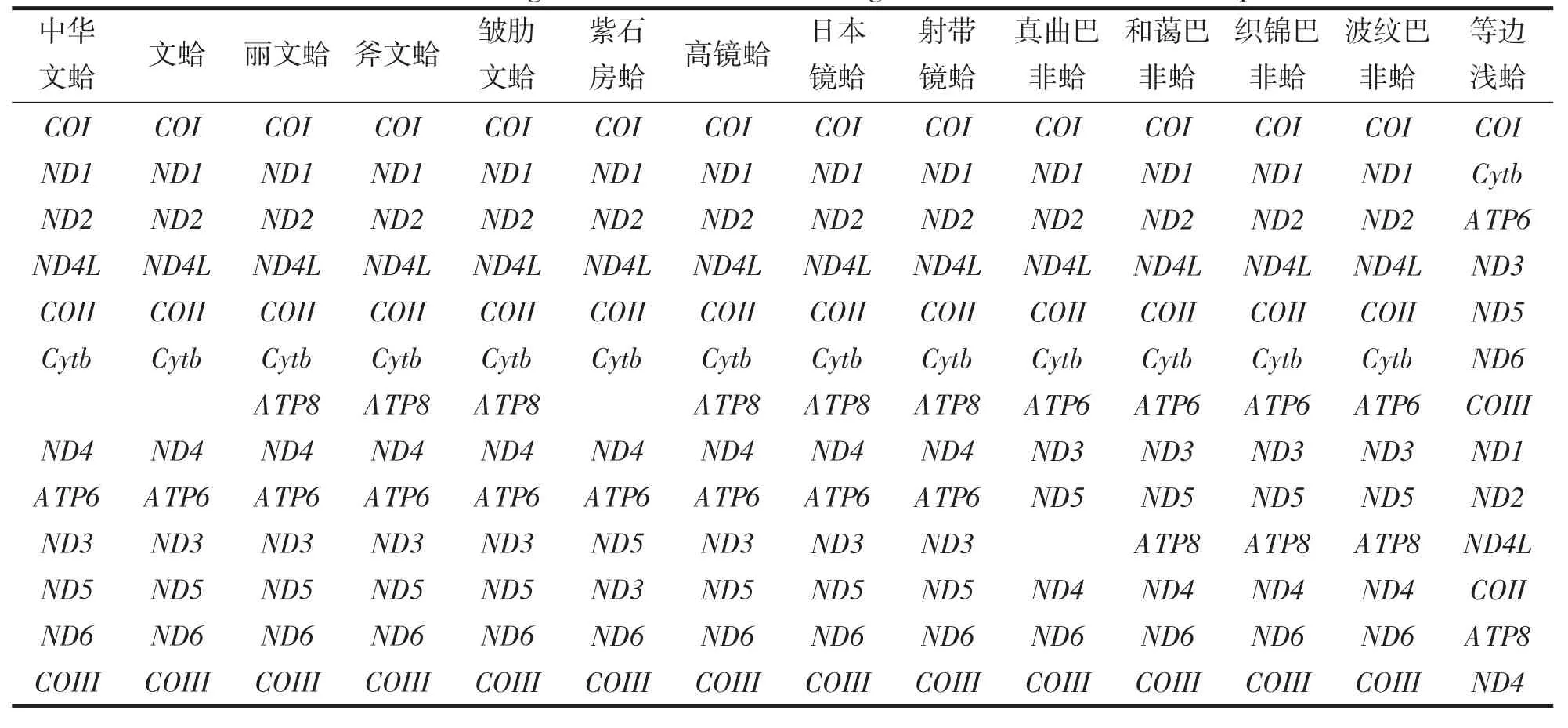
表3 14 种帘蛤科贝类线粒体基因组的基因重排Tab.3 Gene rearrangement of mitochondrial genomes of 14 Veneridae species
2.4 遗传距离
基于线粒体基因组全序列运用MEGA 6 软件算出14 种帘蛤科贝类之间的遗传距离。结果见表4。物种间的遗传距离在0.006~0.615 之间。最大的遗传距离存在于紫石房蛤与和蔼巴非蛤之间,为0.615;最小的遗传距离存在于中华文蛤和文蛤之间,为0.006,说明2 个物种亲缘关系较近。此外可以看出相同属之间的遗传距离较小,不同属之间的遗传距离较大,等边浅蛤与巴非蛤属亲缘关系较近,紫石房蛤与文蛤属物种较近,与后面系统发育树结果一致。
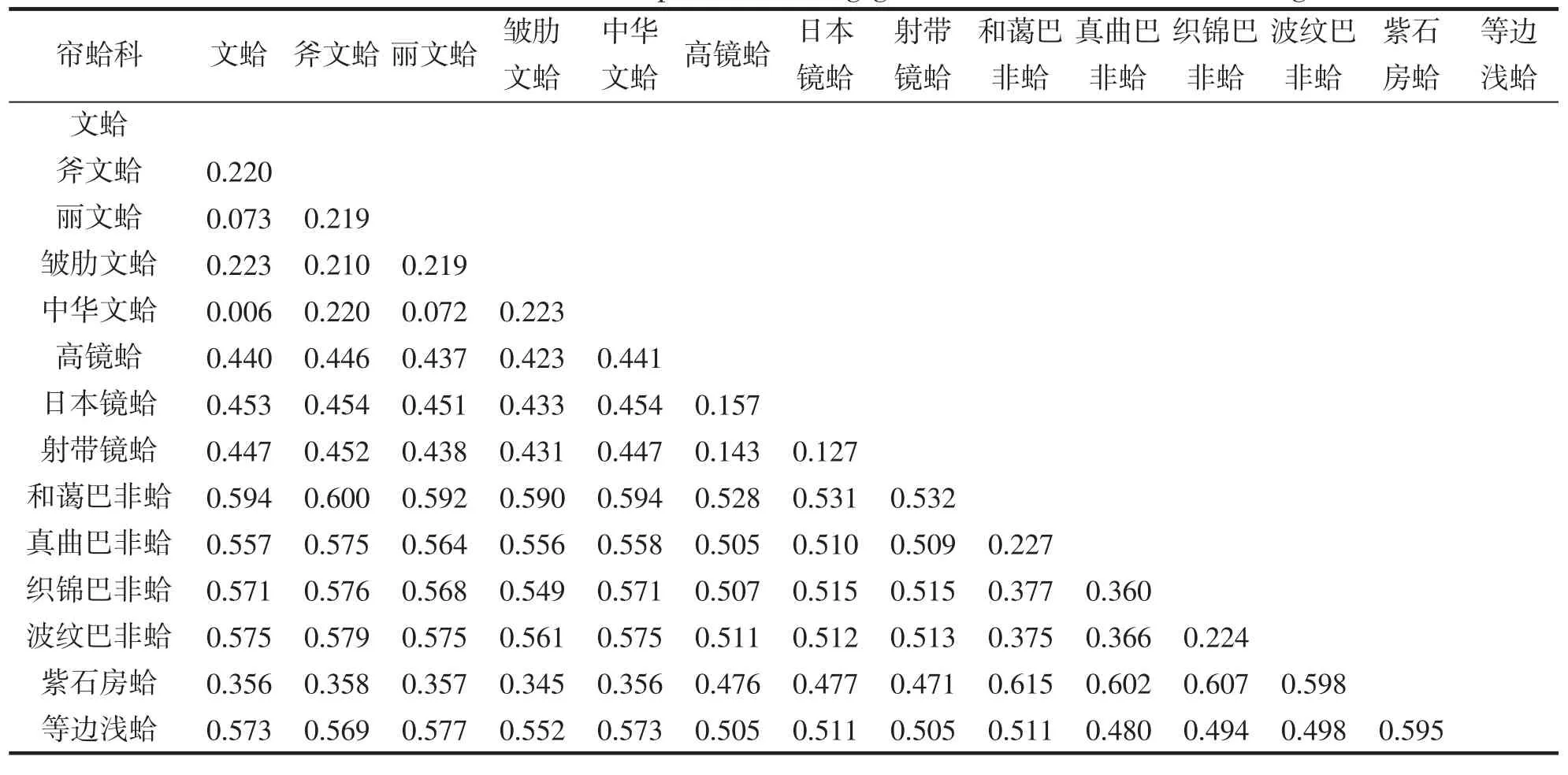
表4 基于线粒体基因组12 个蛋白质编码基因的帘蛤科贝类遗传距离Tab.4 Genetic distance of the 12 protein-coding genes based on mitochondrial genome
2.5 选择压力分析
本研究中选取巴非蛤属的和蔼巴非蛤,真曲巴非蛤,织锦巴非蛤,波纹巴非蛤为代表来研究帘蛤科贝类线粒体基因组选择压力。估算各物种线粒体基因中12 个蛋白质编码基因的同义替换率和非同义替换率以及其比值,构建柱状图。结果如图1 所示,12 种蛋白质编码基因Ka/Ks 均小于1,表明基因受到程度不等负选择(纯化选择)作用。在受到纯化选择压力的所有12 个基因中,ND6 的Ka/Ks 值最大,ND5 基因的Ka/Ks 值最小,其余基因的Ka/Ks 值都在0和1 之间。

图1 4 种帘蛤科贝类蛋白质编码基因Ka/Ks 值的比较Fig.1 Comparison of Ka/Ks ratios in the mitochondrial protein genes of 4 Veneridae shellfishs
2.6 系统发育关系
基于14 个帘蛤科贝类的线粒体全长,用贻贝科的3 个物种作为外群,采用NJ 法构建了帘蛤科贝类的系统发育树。如图2,可见其拓扑结构。帘蛤科贝类分为4 个类群。第一个类群,文蛤亚科的5 个物种与石房蛤属的紫石房蛤聚为一枝,而文蛤亚科的中华文蛤和文蛤首先聚为一枝,再与丽文蛤聚为一枝,再与斧文蛤聚为一枝,最后与皱肋文蛤聚为一枝,第二类群由镜蛤属的3 个物种聚为一枝,日本镜蛤和射带镜蛤先聚成一枝,再与高镜蛤聚为一枝,第三类群由浅蛤属的等边浅蛤自成一枝,与组成第四类群的巴非蛤属的4 个物种聚成一枝,而巴非蛤属内的和蔼巴非蛤,真曲巴非蛤和织锦巴非蛤,波纹巴非蛤,两两聚在一起。
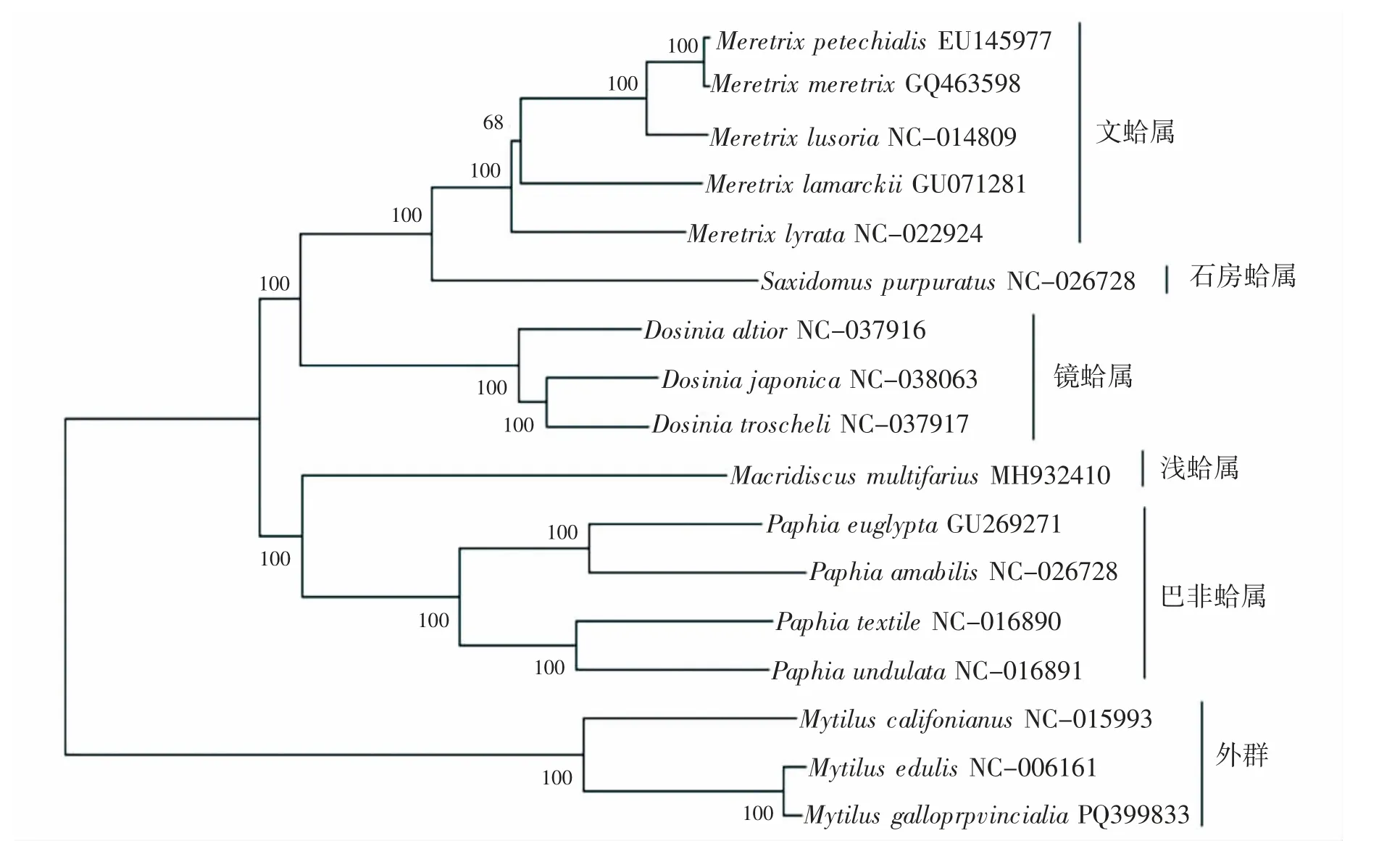
图2 基于12 个蛋白质编码基因构建的NJ 系统发育树Fig.2 The Neighbor-joining phylogenetic tree based on 12 protein-coding genes
3 讨论
研究发现帘蛤科14 种贝类线粒体基因组A+T 含量高于G+C,分析可能是因为12 个蛋白质编码基因,其中除了ND6 由L 链编码,其他11 个基因编码基因,都由H 链编码。而在线粒体基因组在复制过程中,H 链先开始复制,伴随DNA 链复制过程的半不连续所导致的不对称性,L 链在H 链复制了2/3 后才开始复制,在处于单链状态的L 链容易发生碱基突变,从而导致A+T 含量高于G+C 的情况出现[13]。此外在氨基酸密码子组成和数目上看出亲缘关系较近的(同一属内)差异较小,而亲缘关系较远的差异较大。
关于线粒体基因排序,线粒体基因排列作为目前揭示物种之间系统发育关系的有力参考之一,被广泛运用,并且有多种模型被提出来解释线粒体基因的重排[14]。BOORE,et al[15]研究称后生动物线粒体基因由于其基因容量的保守性,所以线粒体基因的排列顺序成为研究后生动物系统发育重要手段之一。软体动物门种类与其他后生动物相比,基因序列容易多变。本研究中帘蛤科14 个物种线粒体基因序列不尽相同,且有基因缺失,即ATP8。ATP8 基因作为ATP 合酶的1 个亚基,参与ATP 合成和细胞的能量产生。文蛤、中华文蛤、紫石房蛤和真曲巴非蛤缺失ATP8,推测原因可能是由于这4 种贝类生活环境水温较低,代谢水平较低,从而在进化过程中导致ATP8 的缺失。此外值得关注的是等边浅蛤基因序列,与其余帘蛤科贝类相比,大为不同。可以看到在等边浅蛤mtDNA 中,发生了滑移重排现象,其中包含Cytb-ATP6-ND3-ND5,ND6-COIII,ND1-ND2-ND4L-COII,ATP8-ND4 这4 个基因框,研究也发现,这种滑移或移位重排可能是由于线粒体基因重组造成的[16]。因此,关于是否由等边浅蛤生活环境造成的基因重排和这种基因重排构建的意义和用途,是仍需要我们探索的。
关于基因同义替换率与非同义替换率的比值,基因突变中通常有3 种分子进化策略导致生物进化。即正选择,中性选择和负选择(纯化作用),而基因突变分为同义突变和非同义突变,因此,当前,主要通过同义替换率和非同义替换率的比值来反映出相对的分子进化率[17-18]。当前主要认为若Ka/Ks>1,则基因受到正选择作用;若Ka/Ks=1,则认为基因受到中性选择作用。若Ka/Ks<1,则认为基因受到负选择作用。本研究中12 种蛋白质编码基因Ka/Ks 值不同且均小于1,表明基因受到程度不等的纯化选择作用。在受到纯化选择压力的所有12 个基因中,ND6 的Ka/Ks 值最大,表明其受到的选择压力最小,而Cytb 基因Ka/Ks 值最小,表明在进化过程中这2 个基因所受的选择压力较大。
而基于线粒体基因组建立的NJ 树和14 个物种基于12 个蛋白质编码基因估算出的遗传距离,得到的结论与庄启谦先生的《中国动物志.软体动物门:双壳纲,帘蛤科》中帘蛤科贝类形态分类结果基本一致。因此研究表明线粒体基因组作为帘蛤科贝类系统进化的研究手段具有一定可靠性。
