茶树基因组与测序技术的研究进展
2021-12-11王鹏杰杨江帆张兴坦叶乃兴
王鹏杰,杨江帆,张兴坦,叶乃兴
茶树基因组与测序技术的研究进展
王鹏杰1,2,杨江帆1,张兴坦1,2*,叶乃兴1*
1.福建农林大学园艺学院,茶学福建省高校重点实验室,福建 福州 350002;2. 中国农业科学院深圳农业基因组研究所,广东 深圳 518120
茶树具有高度杂合、基因组庞大及高度重复等特点,这导致茶树基因组的前期研究进展缓慢。基因组测序技术的迅速发展有力推动茶树基因组的解析与完善。综述了基因组测序技术的发展,将近年来茶树基因组的组装与研究进展按照草图水平、染色体水平和单体型水平进行分类,探讨茶树基因组未来的应用与发展方向,为茶树功能基因组学研究和精确分子育种提供参考。
茶树;测序技术;基因组;染色体水平;单体型
茶树[(L.) O. Kuntze]是我国重要的经济作物,我国是世界上最早栽培茶树和利用茶叶的国家[1]。据统计,茶树已在60多个国家和地区进行商业种植,每年采摘的茶叶超过500万t[2]。目前,全球范围内的消费者每天饮茶超过20亿杯,这些茶产品主要是通过茶树芽叶加工而成的,并富含特征性的次级代谢物,包括儿茶素、茶氨酸、咖啡碱和多种挥发性化合物,有益于人们身体健康[3]。2019年联合国大会(General Assembly of the United Nations,UNGA)将每年的5月21日指定为“国际茶日”,表彰茶在国际社会、经济与文化发展中的重要价值,尤其是其在发展中国家农村减贫和发展中的宝贵作用。然而,与茶在全球经济产业中的巨大贡献相比,茶树的基础生物学研究和育种效率仍较为落后[4],迫切需要高质量的茶树基因组以促进茶树的基础生物学研究和分子育种。而由于茶树庞大的3 Gb大小基因组、超过70%的重复序列含量以及自交不亲和导致的高度杂合特性[5-11],对茶树基因组的测序和组装是巨大挑战。随着基因组测序技术的迅速发展,2017年中国科学院昆明植物研究所的研究团队发布了基于二代测序获得的大叶种云抗10号茶树基因组[5],茶学研究人员开始在茶树全基因组范围内进行深入的基础科学研究。本文主要综述了基因组测序技术及茶树基因组学的研究进展,探讨茶树基因组未来的应用与发展方向,以期为茶树功能基因组学和精确分子育种提供参考。
1 基因组测序技术研究进展
1.1 一代测序技术
DNA测序技术在过去四十多年以来经历了三代革新,极大地推动基因组学的研究进展,为动植物的基础生命科学研究奠定了雄厚基础。第一代测序技术始于Sanger等[12]1977年开创的双脱氧链末端终止法,该方法将附有放射性同位素标记的ddNTP分别混入4个DNA合成反应过程中,以电泳条带的位置确定DNA序列。基于Sanger测序法,ABI公司生产了第一代3730XL测序仪器,其有效读长可以达到1 kb,且每个碱基的准确程度达到99.999%。依靠着一代测序技术的精确性,模式植物拟南芥()的基因组于2000年被解析[13],成功开启了植物基因组学研究的新纪元。2001年在[14]和[15]杂志发表的人类基因组研究成果就是在Sanger测序基础上改进完成的,该成果成为人类生命科学研究的重要丰碑。此后,陆续有不少植物通过一代测序被解析,包括水稻()[16]、杨树()[17]、葡萄()[18]、番木瓜()[19]、高粱()[20]、玉米()[21]等重要植物。虽然一代测序技术获得的序列准确程度极高,但是成本昂贵、低通量、耗时长的缺点制约其进一步发展,在目前的大规模测序中只能成为辅助手段。
1.2 二代测序技术
由于功能基因组学时代的到来和测序技术的巨大进步,454焦磷酸测序[22]、Solexa测序及SOLiD测序等新一代技术兴起,并统称为二代测序技术[23]。二代测序技术依靠着低成本、高通量、少耗时等优势逐渐取代第一代测序技术成为大规模动植物基因组测序的主要技术。尽管二代测序技术的准确性不如一代测序,且获得reads的读长较短,但通过提高测序深度,可以很好的弥补这些缺陷。当前,美国Illumina公司的Solexa技术占据了市场主要份额,其开发的Illumina平台特点为边合成边测序,在DNA合成反应体系中添加接头引物、DNA聚合酶及拥有碱基特异性荧光的dNTP,不同的碱基特异荧光可以区分碱基信号,并根据荧光颜色判断碱基组成类型。此外,通过桥式PCR技术,可对Illumina文库进行双端测序。目前公布的大部分植物基因组均是基于Illumina测序平台完成的,包括黄瓜()[24]、可可树()[25]、白菜()[26]、木豆()[27]、甜橙()[28]、西瓜()[29]等。然而,由于读长短的劣势,二代测序不能较好的组装那些高重复性、高杂合性及高度结构变异的动植物基因组。
1.3 三代测序技术
近年来,随着测序技术进一步发展,被称为单分子实时测序技术的三代测序技术,在动植物测序中大量使用。该测序技术不需要扩增文库,直接对每一个单DNA分子进行测序。三代测序相比于二代测序最典型的特征为超长读长,且耗时更短。然而,其随机错误率较高,准确性不如二代和一代测序技术[30-31]。目前,三代测序以Pacific Biosciences公司的PacBio SMRT测序技术和Oxford Nanopore公司的纳米孔测序技术为主。其中,PacBio SMRT测序技术同样以边测序边合成为原理,DNA分子在测序过程中进入10~50 nm的纳米孔,然后成为模板在DNA聚合酶的帮助下进行复制;带有4种碱基特异荧光的标记基团在与模板配对过程中可显示特异荧光,最后通过分析荧光波长和峰值判断碱基类型。PacBio SMRT测序技术获得的数据随机错误率高,限制了该技术的应用。而在2019年最新发布的PacBio Sequl技术,通过循环一致性测序(Circular consensus sequencing,CCS)模式可产生99.8%的高精确、高保真数据,且平均读长可达到13.5 kb[32]。纳米孔测序技术的测序原理则是基于单DNA分子穿过生物纳米孔时由于碱基类型差异引起电流强度变化,以此判断碱基组成类型。最近3年的植物基因组测序大多选择了三代测序技术[33-35],包括最近发表的多个茶树染色体级别参考基因组[7-10]。由于三代测序技术超长的reads,使其在超大、高重复、高杂合的基因组测序与组装层面上具有显著优势,随着价格的不断下降,将在后续动植物基因组组装中大量运用。
1.4 辅助组装技术
无论是几代基因组测序技术,最后获得的reads都需要进行组装。而之前的植物基因组组装大都只能组装至contigs和scaffolds层次,或通过对子代群体测序构建遗传图谱以进一步将contigs或scaffolds挂载到接近染色体级别的基因组。但是由于技术的限制,其遗传图谱的标记一般很难满足长度较短的contigs或scaffolds的挂载需求,导致挂载率和准确性较低。近年来发展起来的新一代基因组辅助组装技术很好地解决了这些问题,其中应用最广的当属高通量染色体构象捕获(High-through chromosome conformation capture,Hi-C)技术[36]。Hi-C技术通过高通量测序在较短时间内获得大量染色质DNA在空间上的互作关系。该技术辅助基因组组装的原理是界定不同染色体间存在一定的“疆域”,而单条染色体内的互作频率应大于不同染色体间的互作频率,且染色体内近端的互作频率也应大于远端的,由此可初步将获得的原始基因组装配分配到不同染色体上。此外,还有BioNano光学图谱技术,其原理主要是通过添加多个基因组标记位点,将DNA分子在纳米孔上线性展开,并通过荧光成像扫描位点转化成基因组物理图谱信息以改善基因组质量[37-38]。相比于BioNano光学图谱技术,Hi-C技术在植物基因组辅助组装上应用更为广泛,不仅节约成本与时间,且挂载率和准确性极高。此外,由于部分植物的杂合度极高,其测定的基因组信息折叠了大量的遗传变异信息,而基于Hi-C数据的多种算法程序可较好地构建二倍体或多倍体植物的单体型基因组,例如FALCON-Phase[39]和ALLHiC等[40]。FALCON-Phase可通过整合二倍体植物三代测序和Hi-C数据,将初级装配构建为高质量的单体型基因组装配体。ALLHiC通过“修剪”步骤,删除等位基因之间的链接,即单体型之间的链接,不仅可以构建高度杂合的二倍体植物的单体型基因组,也适用于多倍体基因组的单体型定相,并且已成功应用于几种植物的基因组组装中,包括同源四倍体[33]和同源八倍体[41]的甘蔗基因组、同源四倍体的紫花苜蓿基因组[35]和榕树基因组[34]。
2 茶树基因组研究进展
茶树高质量基因组有助于茶树的起源驯化与生理特性研究,能加速理想性状茶树种质的分子育种。如图1所示,自2017年至今,已正式发表了多篇茶树基因组相关的高水平论文[3,5-10],其中持续提高的茶树基因组质量、不断深入的茶树生物学问题,均有力地促进了科研工作者对茶树基础理论研究的整体认识。正如自21世纪元年宏伟的人类基因组初次公布至今,基因组带给人类对生命奥秘的认识已经大大超出了预期。
2.1 草图水平的茶树基因组
2017年,中国科学院昆明植物研究所的研究团队基于二代测序技术率先公布了大叶种茶树云抗10号的基因组序列[5],拉开了茶树基因组学研究的帷幕。云抗10号的基因组大小为3.02 Gb,含有36 951个注释编码蛋白,其中contig N50为19.96 kb,scaffold N50为0.45 Mb。该研究首次观察到茶树中的重复序列含量在基因组中占极高比例,几乎达到80.9%,并认为这可能是由于茶树在长达5 000万年的时间维度内长末端重复序列反转录转座子家族大量爆发扩张,而缺少相应的DNA删除机制,使得茶树基因组庞大且重复序列含量极高。此外,还发现云抗10号部分与茶叶风味品质相关的基因家族发生扩张,例如类黄酮、萜类等化合物合成相关基因。该研究观察到茶树的咖啡碱合成途径相比于可可和咖啡,存在独立且迅速的驯化过程。这些结果可能影响茶树的环境适应性和制茶品质。2018年,安徽农业大学的茶学团队基于二代Illumina测序为主、三代PacBio测序补洞的方法测序获得了茶树小叶种品种舒茶早的基因组序列[6]。该基因组基于scaffold水平的大小为3.14 Gb,具有33 932个注释编码蛋白,其中contig N50为67.07 kb,scaffold N50为1.39 Mb,重复序列的基因组占比为64%,组装质量较云抗10号基因组有较大提升。该研究发现,茶树的祖先种与猕猴桃的物种分化时间可能在8 000万年前。进一步的分析发现大叶种和小叶种可能是在38至154万年前从其原始祖先种中分化而来,两者具有极高的共线性。在舒茶早基因组中观察到两次全基因组复制事件,其中近期的一次复制事件影响了大量次生代谢相关基因的扩张,尤其是与儿茶素合成相关的基因,这可能可以解释栽培茶树儿茶素组分的高含量。该研究还挖掘了影响茶氨酸合成的关键酶基因并验证功能。此外,通过比较基因组学发现,在茶叶香气组成中占重要位置的萜烯类化合物,其合成酶基因也经历显著扩增。后续该团队进一步改善舒茶早基因组的注释质量[42],并整合茶树基因组、转录组、代谢组等数据开发TPIA数据库(http://tpia.teaplant.org/index.html)[43],有力推进了茶树的基础研究与数据共享。
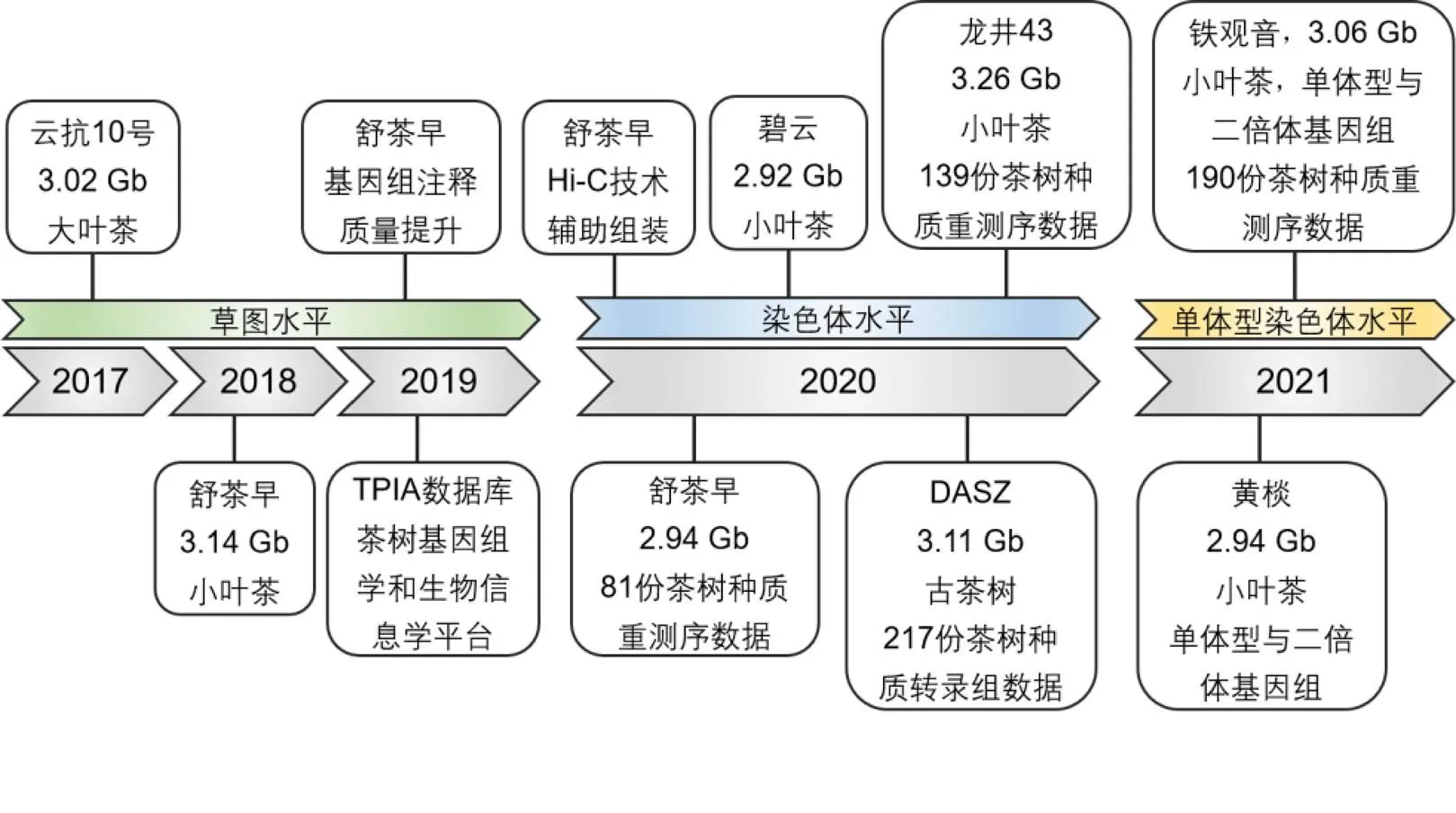
图1 茶树基因组研究进展时间线
2.2 染色体水平的茶树基因组
随着三代测序和Hi-C技术的成熟和价格的下降,2020年度茶学领域涌现了多个组装到染色体水平的高质量茶树基因组。中国农业科学院茶叶研究所的种质资源团队采用Hi-C技术将之前公布的舒茶早基因组组装至染色体水平[3],scaffold N50达到218.1 Mb。该研究重点分析了茶树的全基因组复制情况,发现茶树在1.466~1.527亿年前及5.89~6.17千万年前分别经历了一次古六倍体事件和古四倍体事件。QTL数据的染色体定位发现部分儿茶素相关QTL在古四倍体化事件后发生分化。
安徽农业大学的茶学团队进一步在之前组装的舒茶早草图基因组基础上,通过PacBio SMRT和Hi-C技术构建了染色体级别的茶树基因组[10],该基因组基于contig水平的大小为2.94 Gb,具有50 525个注释编码蛋白,其中contig N50为600.46 kb,scaffold N50为167 Mb,显著提升了茶树基因组的组装质量。该研究发现了2.55 Gb的重复序列,达到整个基因组的86.87%。首次系统鉴定了茶树基因组的杂合区域,该区域占整个基因组的18.8%,包含3 440个基因,参与多个生物学过程。研究还揭示了萜类合成酶基因家族在茶树染色体上以基因簇形式分布并在近期发生串联重复事件。最重要的是,通过81份国内外茶树种质的重测序,发现了栽培种和野生种茶树的基因变异与驯化足迹,开启了茶树起源与驯化研究的新阶段。与此同时,华南农业大学的研究团队也公布了小叶种碧云的染色体级别基因组序列[7],该基因组基于scaffold水平的大小为2.92 Gb,共有40 812个高质量注释基因,其中contig N50为625.11 kb,scaffold N50为195.68 Mb,重复序列占基因组的74.13%,并着重揭示了LTR逆转录转座子在茶树中的起源、进化与变异。此外,中国农业科学院茶叶研究所的研究团队也公布了优质绿茶适制品种龙井43的染色体级别基因组序列[8],该基因组共3.26 Gb,编码33 556个注释蛋白,contig N50为271.33 kb,scaffold N50为143.85 Mb。作为抗逆性极强的茶树品种,龙井43基因组的抗逆相关基因经历正选择,抗逆、品质、自交不亲和等相关基因经历扩张。139份国内外代表种质的重测序表明茶树的驯化过程中,小叶种茶树的萜类相关基因与抗病基因受到相对于大叶种茶树更强的人工选择。
对古茶树的研究有利于揭示茶树的起源与驯化。华中农业大学的研究团队也于2020年7月公布了取自云南省野外深山的古茶树DASZ基因组序列[9],该基因组为3.11 Gb,编码33 021个注释蛋白,contig N50为2.59 Mb。通过对主要产茶省份217份茶树资源的转录组测序,发现古茶树与栽培种茶树并未有显著分化,且论证了福鼎大白茶与铁观音等品种作为中国茶树骨干亲本的重要地位,并通过全基因组关联分析鉴定了多个与儿茶素合成有关的遗传位点与相关基因。
福建农林大学与中国农业科学院农业基因组研究所的研究团队于2021年5月和7月先后发表了乌龙茶品种黄棪[44]和铁观音[45]的染色体级别基因组。黄棪茶树基因组大小为2.94 Gb,杂合度为2.79%~3.40%,contig N50达到2.61 Mb,注释获得43 779个蛋白质编码基因,70.75%的黄棪基因组序列被注释为重复序列。研究发现黄棪与古茶树DASZ之间具有最多的结构变异注释基因,与绿茶品种舒茶早及龙井43之间的结构变异注释基因与香气途径相关,表明结构变异可能影响黄棪的高香品种特性。结合结构变异、转录组和挥发物测定结果发现萜类合成酶(Terpene synthase,TPS)家族基因的结构变异、广泛且特异地高表达是黄棪品种高香特性的分子基础。基于黄棪的高质量基因组,该团队还探究了茶树在低温胁迫的染色质可及性、转录与翻译图谱[46],有助于阐明茶树乃至植物如何灵活地协调染色质、转录和翻译的效应以应对低温胁迫。铁观音茶树基因组大小为3.06 Gb,contig N50为1.94 Mb,BUSCO完整性达到93.7%,注释获得42 825个蛋白质编码基因,并确定了2.39 Gb的重复序列,占基因组大小的78.2%。重要的是,通过190份茶树种质的基因组学分析揭示了大叶和小叶茶的独立进化与平行驯化历史,发现了广泛的种内和种间渗入,并且揭示茶树潜在的“绿色革命”基因和,这有助于现今茶树品种的遗传多样性和不同树型的形成。
2.3 单体型解析的茶树基因组
茶树和其他植物的二倍体或多倍体基因组往往由两套或多套染色体组组成。值得注意的是,大多数植物二倍体参考基因组的组装结果混杂了双亲等位基因组的嵌合序列,尽管这样的组装便于比较和分析数据,但都忽略了可能具有潜在生物学功能并影响高度杂合基因组质量的等位基因变异[40]。单体型解析的基因组组装是植物基因组学领域的新趋势,有助于植物杂种优势与进化研究,并为准确、可靠的基因组编辑提供了坚实的基础,但是具体的分型方法仍是一个巨大的挑战[47]。
茶树具有自交不亲和特性,导致其基因组高度杂合并存在大量等位基因变异。华中农业大学的研究团队对茶树品种福鼎大白茶135个精细胞进行分离与全基因组测序,基于古茶树DASZ的二倍体基因组数据进行单体型分型[48],并系统地分析了被誉为“华茶1号”的福鼎大白茶与中国各省份106份茶树资源的亲缘关系,为茶树的品种选育以及基因表达调控研究提供了新的见解。
二倍体植物两套单体型基因组的等位基因变异可能在表型调控、杂种优势和进化中起重要作用。前人的研究支持杂交水稻具有杂种优势的关键因素是具有高转录活性和显性表达的等位基因[49-50]。通过结合三代HiFi数据、Hi-C技术、Hifiasm及ALLHiC程序,福建农林大学与中国农业科学院深圳农业基因组的研究团队首次公布了高杂合性茶树的单体型染色体级别基因组[44-45]。其中,茶树品种黄棪的两套单体型基因组总共包含30条假染色体,单体型A长度为2.90 Gb,单体型B为2.97 Gb。两组单体型基因组间存在大量遗传变异,包括2 357万个SNP,114万个插入和113万个缺失。59.38%的编码基因可定义为等位基因,平均相似度为92.60%。茶树品种铁观音的两套单体型基因组长度分别为3.06 Gb和2.92 Gb,其中34.46%的注释基因可定义为等位基因,并发现1 528个表达模式一致性的和386个不一致的等位特异性表达基因。这表明在茶树长期的繁殖培育过程中,显性效应在克服突变负荷中起着重要作用。
3 总结与展望
茶树基因组学研究的重大进展,有效推动了茶树功能基因组学与重要农艺性状基因的相关研究,也为茶树的起源与演化等基础生物学问题提供借鉴,并深化了人们对茶的基础认识与加工利用。在未来的茶树基因组学研究与应用中,可以关注以下几个方向:
(1)茶树具有高度的杂合性,单体型水平的分型基因组更能代表茶树的真实基因信息,后续的研究可通过单体型基因组数据进一步挖掘茶树的等位基因差异对表型和代谢产物的影响。值得关注的是,由于亲本基因组数据资源的缺失,茶树杂种优势遗传机理的研究一直较为落后。最近公布的铁观音和黄棪基因组可以成为茶树杂种优势研究的模式数据。铁观音和黄棪是乌龙茶育种的核心亲本,具有互补的优质性状。铁观音具有天然“兰花香”和独特“观音韵”,但存在萌芽期晚、产量较低及制优率偏低等不足之处;而黄棪具有高香、萌芽期早、产量较高、制优率高等优点,是国家茶树品种区域试验的乌龙茶对照种。以铁观音和黄棪为亲本创新衍生了一系列茶树优良品种[51-53],包括金观音(国审2002017)[54-55]、黄观音(国审2002015)[56]、金牡丹(国品2010024)、紫牡丹(国品2010026)、黄玫瑰(国品2010025)、春兰(国品2010016)、黄奇(国审2002018)[57]、瑞香(国品2010017)[58]、紫玫瑰(闽审2005003)、春闺(闽审2015001)[59]、凤圆春(闽审99001)、鸿雁1号(国品2010022)、鸿雁12号(国品2010020)、鸿雁13号(国品2014010)等国家级或省级良种,还包括春桃香和金茗早[60]等多个新品系。因此,铁观音和黄棪家系是乌龙茶品种资源中最为重要的组成群体。基于已发表的铁观音和黄棪高质量二倍体与单体型基因组,后续可以定量该后代杂交群体的亲本等位基因杂种优势,挖掘不同杂种优势代谢物的等位基因调控,为茶树高香优质杂交品种的选育提供理论依据。
(2)由于不同茶树种质之间特有的遗传性状,单一品种的基因组已经无法代表茶树所有的遗传信息。通过重新整合已经公布的云抗10号、舒茶早、龙井43、碧云、古茶树DASZ、铁观音及黄棪基因组原始数据,并且补测不同茶区的代表性茶树品种基因组数据,共同构建泛基因组(Pan-genome),将是茶树基因组未来的发展趋势。基于代表性茶树种质的泛基因组可以全面捕捉茶树的遗传信息,为茶树功能基因组学研究和精确分子育种提供宝贵资源。
(3)基于高质量的茶树基因组数据进行更为前沿的组学分析。例如:三维基因组学、单细胞系列组学、空间组学等。这些数据可以让我们在染色质三维结构层面、单细胞层面和空间层面上挖掘细致深入的转录调控事件,从而全面地解析茶树的基础生物学问题。
(4)栽培茶树的起源与驯化一直是众多研究者关注的焦点,尽管已经有了一些文献学、语音学、考古学及遗传学上的认识,但仍需要更多的证据去证明茶树祖先类型、起源地点、时间以及驯化史等[61]。随着测序技术的飞速发展,如今二代测序的每Gb数据价格已经降到了50元以内[62]。因此,可以通过整合目前已经发表的多个茶树基因组以及数百份茶树种质重测序数据,并且补测更多的代表性茶树种质与近缘种质资源的重测序数据,包括其他国家的茶树种质在内,从而在更大样本的基因组水平上提供茶树起源与驯化的遗传学证据。
(5)基于茶树全基因组信息与重要农艺性状的全基因组关联分析(GWAS)是今后重要的研究应用方向。尽管目前已经通过简化基因组测序等获得的相关数据对少数性状进行了初步定位[63-66],仍需要通过结合覆盖度更广的基因组重测序数据与更全面的性状调查数据,在高精度的茶树基因组上进行重要农艺性状的关键位点鉴定,为后续茶树遗传转化体系化时代的到来提供分子育种的核心基因资源。
[1] 叶乃兴. 茶学研究法[M]. 北京: 中国农业出版社, 2011.
Ye N X. Research methods of tea science [M]. Beijing: China Agriculture Press, 2011.
[2] Drew L. The growth of tea [J]. Nature, 2019, 566: s2-s4.
[3] Chen J D, Zheng C, Ma J Q, et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant [J]. Horticulture Research, 2020, 7: 63. doi:10.1038/s41438-020-0288-2.
[4] Xia E H, Tong W, Wu Q, et al. Tea plant genomics: achievements, challenges and perspectives [J]. Horticulture Research, 2020, 7: 7. doi: 10.1038/s41438-019-0225-4.
[5] Xia E H, Zhang H B, Sheng J, et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis [J]. Molecular Plant, 2017, 10(6): 866-877.
[6] Wei C L, Yang H, Wang S, et al. Draft genome sequence ofvar.provides insights into the evolution of the tea genome and tea quality [J]. Proceedings of the National Academy of Sciences of the United States of America, 2018, 115(18): E4151-E4158.
[7] Zhang Q J, Li W, Li K, et al. The chromosome-level reference genome of tea tree unveils recent bursts of non-autonomous LTR retrotransposons to drive genome size evolution [J]. Molecular Plant, 2020, 13(7): 935-938.
[8] Wang X, Feng H, Chang Y, et al. Population sequencing enhances understanding of tea plant evolution [J]. Nature Communications, 2020, 11(1): 4447.doi: 10.1038/s41467-020-18228-8.
[9] Zhang W Y, Zhang Y J, Qiu H J, et al. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties [J]. Nature Communication, 2020, 11(1): 3719.doi: 10.1038/s41467-020-17498-6.
[10] Xia E H, Tong W, Hou Y, et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into genome evolution and adaptation of tea plants [J]. Molecular Plant, 2020, 13(7): 1013-1026.
[11] Jia X, Zhang W, Fernie A R, et al.(Tea) [J]. Trends in Genetics, 2021, 37(1): 201-202.
[12] Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors [J]. Proceedings of the National Academy of Sciences of the United States of America, 1977, 74(12): 5463-5467.
[13] Arabidopsis G I. Analysis of the genome sequence of the flowering plant[J]. Nature, 2000, 408(6814): 796-815.
[14] Venter J C, Adams M D, Myers E W, et al. The sequence of the human genome [J]. Science, 2001, 291(5507): 1304-1351.
[15] Lander E S, Linton L M, Birren B, et al. Initial sequencing and analysis of the human genome [J]. Nature, 2001, 409(6822): 860-921.
[16] Yu J, Hu S, Wang J, et al. A draft sequence of the rice genome (L. ssp.) [J]. Science, 2002, 296(5565): 79-92.
[17] Tuskan G A, Difazio S, Jansson S, et al. The genome of black cottonwood,(Torr. & Gray) [J]. Science, 2006, 313(5793): 1596-1604.
[18] Jaillon O, Aury J, Noel B, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla [J]. Nature, 2007, 449(7161): 463-467.
[19] Ming R, Hou S, Feng Y, et al. The draft genome of the transgenic tropical fruit tree papaya (Linnaeus) [J]. Nature, 2008, 452(7190): 991-996.
[20] Paterson A H, Bowers J E, Bruggmann R, et al. The Sorghum bicolor genome and the diversification of grasses [J]. Nature, 2009, 457(7229): 551-556.
[21] Schnable P S, Ware D, Fulton R S, et al. The B73 maize genome: complexity, diversity, and dynamics [J]. Science, 2009, 326(5956): 1112-1115.
[22] Margulies M, Egholm M, Altman W E, et al. Genome sequencing in microfabricated high-density picolitre reactors [J]. Nature, 2005, 437(7057): 376-380.
[23] Levy S E, Myers R M. Advancements in next-generation sequencing [J]. Annual Review of Genomics and Human Genetics, 2016, 17: 95-115.
[24] Huang S, Li R, Zhang Z, et al. The genome of the cucumber,L. [J]. Nature Genetics, 2009, 41(12): 1275-1281.
[25] Argout X, Salse J, Aury J, et al. The genome of Theobroma cacao [J]. Nature Genetics, 2011, 43(2): 101-108.
[26] Wang X, Wang H, Wang J, et al. The genome of the mesopolyploid crop species[J]. Nature Genetics, 2011, 43(10): 1035-1039.
[27] Varshney R K, Chen W, Li Y, et al. Draft genome sequence of pigeonpea (), an orphan legume crop of resource-poor farmers [J]. Nature Biotechnology, 2011, 30(1): 83-89.
[28] Xu Q, Chen L L, Ruan X, et al. The draft genome of sweet orange () [J]. Nature Genetics, 2013, 45(1): 59-92.
[29] Guo S, Zhang J, Sun H, et al. The draft genome of watermelon () and resequencing of 20 diverse accessions [J]. Nature Genetics, 2013, 45(1): 51-58.
[30] 杨官品, 郭栗. 基因组的测序技术及其发展趋势[J]. 中国海洋大学学报(自然科学版), 2017, 47(s1): 48-57.
Yang G P, Guo L. Technologies available for genome sequencing and their advancements [J]. Periodical of Ocean University of China, 2017, 47(s1): 48-57.
[31] Levy S E, Boone B E. Next-generation sequencing strategies [J]. Cold Spring Harbor Perspectives in Medicine, 2019, 9(7): a25791.doi: 10.1101/cshperspect.a025791.
[32] Wenger A M, Peluso P, Rowell W J, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome [J]. Nature Biotechnology, 2019, 37(11): 1155-1162.
[33] Zhang J S, Zhang X T, Tang H B, et al. Allele-defined genome of the autopolyploid sugarcaneL. [J]. Nature Genetics, 2018, 50(11): 1565-1573.
[34] Zhang X, Wang G, Zhang S, et al. Genomes of the banyan tree and pollinator wasp provide insights into fig-wasp coevolution [J]. Cell, 2020, 183(4): 875-889.
[35] Chen H T, Zeng Y, Yang Y Z, et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa [J]. Nature Communications, 2020, 11(1): 2494.doi: 10.1038/s41467-020-16338-x.
[36] Burton J N, Adey A, Patwardhan R P, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions [J]. Nature Biotechnology, 2013, 31(12): 1119-1125.
[37] Dong Y, Xie M, Jiang Y, et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat () [J]. Nature Biotechnology, 2013, 31(2): 135-141.
[38] 陈萍. BioNano图谱数据建模及光学图谱在水稻基因组的应用研究[D]. 北京: 中国科学院大学, 2019.
Chen P. BioNano data modeling and application research of optical atlas in rice genome [D]. Beijing: Chinese Academy of Sciences University, 2019.
[39] Kronenberg Z N, Rhie A, Koren S, et al. Extended haplotype-phasing of long-read de novo genome assemblies using Hi-C [J]. Nature Communications, 2021, 12(1): 1935.doi: 10.1038/s41467-020-20536-y.
[40] Zhang X T, Wu R X, Wang Y B, et al. Unzipping haplotypes in diploid and polyploid genomes [J]. Computational and Structural Biotechnology Journal, 2019, 18: 66-72.
[41] Zhang X T, Zhang S C, Zhao Q, et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data [J]. Nature Plants, 2019, 5(8): 833-845.
[42] Xia E, Li F, Tong W, et al. The tea plant reference genome and improved gene annotation using long-read and paired-end sequencing data [J]. Scientific Data, 2019, 6(1): 122. doi: 10.1038/s41597-019-0127-1.
[43] Xia E H, Li F D, Tong W, et al. Tea Plant Information Archive (TPIA): A comprehensive genomics and bioinformatics platform for tea plant [J]. Plant Biotechnology Journal, 2019, 17(10): 1938-1953.
[44] Wang P, Yu J, Jin S, et al. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome [J]. Horticulture Research, 2021, 8: 107. doi: 10.1038/s41438-021-00542-x.
[45] Zhang X. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant,[J]. Nature Genetics, 2021. doi: 10.1038/s41588-021-00895-y.
[46] Wang P, Jin S, Chen X, et al. Chromatin accessibility and translational landscapes of tea plants under chilling stress [J]. Horticulture Research, 2021, 8: 96.doi: 10.1038/s41438-021-00542-x.
[47] Zhou Q, Tang D, Huang W, et al. Haplotype-resolved genome analyses of a heterozygous diploid potato [J]. Nature Genetics, 2020, 52(10): 1018-1023.
[48] Zhang W, Luo C, Scossa F, et al. A phased genome based on single sperm sequencing reveals crossover pattern and complex relatedness in tea plants [J]. Plant Journal, 2020, 105(1): 197-208.
[49] Huang X H, Yang S H, Gong J Y, et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis [J]. Nature Communications, 2015, 6: 6258.doi: 10.1038/ncomms7258.
[50] Shao L, Xing F, Xu C, et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis [J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(12): 5653-5658.
[51] Zheng Y C, Wang P J, Chen X J, et al. Transcriptome and metabolite profiling reveal novel insights into volatile heterosis in the tea plant () [J]. Molecules, 2019, 24(18): 3380.doi: 10.3390/molecules24183380.
[52] 叶乃兴. 乌龙茶种质资源的利用与品种创新[J]. 福建茶叶, 2006(3): 2-4.
Ye N X. Utilization of oolong tea germplasm resources and cultivar innovation [J]. Tea In Fujian, 2006(3): 2-4.
[53] Zeng L, Zhou X, Su X, et al. Chinese oolong tea: an aromatic beverage produced under multiple stresses [J]. Trends in Food Science and Technology, 2020, 106: 242-253.
[54] 王让剑, 杨军, 孔祥瑞, 等. 利用SSR标记分析金观音(半)同胞茶树品种遗传差异[J]. 茶叶科学, 2017, 37(2): 139-148.
Wang R J, Yang J, Kong X R, et al. Genetic analysis of full- and half-sib families of tea cultivar jinguanyin based on SSR molecular markers [J]. Journal of Tea Science, 2017, 37(2): 139-148.
[55] 姚雪倩, 郑玉成, 王鹏杰, 等. 金观音与其亲本差异基因表达的遗传分析[J]. 福建农林大学学报(自然科学版), 2019, 48(2): 155-160.
Yao X Q, Zheng Y C, Wang P J, et al. Genetic analysis of differential gene expression between Jinguanyin and its parents [J]. Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2019, 48(2): 155-160.
[56] 郭吉春, 杨如兴, 张文锦, 等. 茶树杂交种金观音与黄观音的选育及应用[J]. 贵州科学, 2008, 26(2): 20-24.
Guo J C, Yang R X, Zhang W J, et al. Breeding and application of two tea hybrid Jinguanyin and Huangguanyin [J]. Guizhou Science, 2008, 26(2): 20-24.
[57] 郭吉春, 叶乃兴, 何孝延. 茶树杂交一代展叶期的遗传变异[J]. 茶叶科学, 2004, 24(4): 255-259.
Guo J C, Ye N X, He X Y. Genetic variation in the leaf-expansion period of the first hybrid generation tea plants [J]. Journal of Tea Science, 2004, 24(4): 255-259.
[58] 陈荣冰, 黄福平, 陈常颂, 等. 高香型优质乌龙茶新品系瑞香选育简报[J]. 茶叶科学, 2004, 24(1): 29-32.
Chen R B, Huang F P, Chen C S, et al. Breeding report on strong aroma and good quality newly bred oolong variety Rui xiang [J]. Journal of Tea Science, 2004, 24(1): 29-32.
[59] 钟秋生, 林郑和, 陈常颂, 等. “春闺”绿茶香气成分鉴定分析[J]. 茶叶通讯, 2021, 48(1): 33-39.
Zhong Q S, Lin Z H, Chen C S, et al. Identification and analysis of aroma components in Chungui green tea [J]. Journal of Tea Communication, 2021, 48(1): 33-39.
[60] Chen X, Wang P, Zheng Y, et al. Comparison of metabolome and transcriptome of flavonoid biosynthesis pathway in a purple-leaf tea germplasm Jinmingzao and a green-leaf tea germplasm Huangdan reveals their relationship with genetic mechanisms of color formation [J]. International Journal of Molecular Sciences, 2020, 21(11): 4167. doi: 10.3390/ijms21114167.
[61] 张文驹, 戎俊, 韦朝领, 等. 栽培茶树的驯化起源与传播[J]. 生物多样性, 2018, 26(4): 357-372.
Zhang W J, Rong J, Wei C L, et al. Domestication origin and spread of cultivated tea plants [J]. Biodiversity Science, 2018, 26(4): 357-372.
[62] 唐蝶, 周倩. 植物基因组组装技术研究进展[J]. 生物技术通报, 2021, 37(6): 1-12.
Tang D, Zhou Q. Research advances in plant genome assembly [J]. Biotechnology Bulletin, 2021, 37(6): 1-12.
[63] Ma J, Yao M, Ma C, et al. Construction of a SSR-based genetic map and identification of QTLs for catechins content in tea plant () [J]. PLoS One, 2016, 9(3): e93131.doi: 10.1371/journal.pone.0093131.
[64] 李小杰, 马建强, 姚明哲, 等. 茶氨酸合成酶基因的SNP挖掘和遗传定位[J]. 茶叶科学, 2017, 37(3): 251-257.
Li X J, Ma J Q, Yao M Z, et al. SNP detection and mapping of theanine synthetase gene in tea plant [J]. Journal of Tea Science, 2017, 37(3): 251-257.
[65] Xu L, Wang L, Wei K, et al. High-density SNP linkage map construction and QTL mapping for flavonoid-related traits in a tea plant () using 2b-RAD sequencing [J]. BMC Genomics, 2018, 19(1): 955.doi: 10.1186/s12864-018-5291-8.
[66] Fang K, Xia Z, Li H, et al. Genome-wide association analysis identified molecular markers associated with important tea flavor-related metabolites [J]. Horticulture Research, 2021, 8(1): 42.doi: 10.1038/s41438-021-00477-3.
Research Advance of Tea Plant Genome and Sequencing Technologies
WANG Pengjie1,2, YANG Jiangfan1, ZHANG Xingtan1,2*, YE Naixing1*
1. College of Horticulture, Fujian Agriculture and Forestry University, Key Laboratory of Tea Science at Universities in Fujian, Fuzhou 350002, China; 2. China Agricultural Genome Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China
The tea plant has the characteristics of high heterozygosity, large genome and high duplication, which has led to the slow progress of the preliminary research on the tea plant genomes. The rapid development of genome sequencing technologies has strongly promoted the deciphering and improvement of the tea plant genomes. This article reviewed the development of genome sequencing technologies, and classified the assembly and research progress of tea plant genomes in recent years according to the draft level, chromosome level and haplotype level. By discussing the future application and development direction of tea plant genomes, it provided a reference for the functional genomics research and precision molecular breeding in tea plants.
, sequencing technology, genome, chromosome level, haplotype
S571.1;Q52
A
1000-369X(2021)06-743-10
2021-07-27
2021-08-31
福建省“2011协同创新中心”中国乌龙茶产业协同创新中心专项(闽教科〔2015〕75号)、福建农林大学茶产业链科技创新与服务体系建设项目(K1520005A01)
王鹏杰,男,博士,主要从事茶树遗传育种与生物技术研究。*通信作者:zhangxingtan@caas.cn;ynxtea@126.com
(责任编辑:赵锋)
