IL-17在心肌缺血再灌注损伤中的研究进展
2021-12-10胡孟芬宓宝斌
胡孟芬 宓宝斌
滨州医学院附属医院老年医学科,山东滨州 256600
心肌梗死是全球主要的病死原因之一[1],早期再灌注治疗对于减小心肌梗死面积和改善临床预后至关重要[2-3],但同时,再灌注本身也会造成额外的心肌损伤,这一过程称为缺血再灌注损伤(ischemia reperfusion injury,IRI)[4]。成功再灌注的患者最终心肌坏死面积大约50%源于再灌注损伤[5]。因此,预防和治疗心肌IRI成为心肌IRI保护领域里的研究热点。在IRI的机制中,炎症反应扮演着重要的角色,炎症介质如TNF、IL-1β、IL-6等显著增加[6]。白介素-17(IL-17)是一种促炎因子,参与了炎症反应及自身免疫性疾病的病理生理过程[7]。越来越多的研究表明,IL-17在心肌IRI中发挥着重要的作用。深入研究IL-17在心肌IRI中的作用机制可以为心肌保护寻找新的治疗靶点提供理论基础。现对这一方面的研究做一综述。
1 心肌IRI的机制
心肌IRI可能的机制主要包括细胞内和线粒体Ca2+超载、心肌梗死部位的炎症反应、氧化应激、补体激活和凋亡、自噬等。线粒体通透性转换孔(mPTP)的开放、Toll样受体及microRNA等也与心肌IRI有关。心肌缺血后,再灌注治疗可以诱导补体活化,引起炎症细胞(包括中性粒细胞、淋巴细胞、树突状细胞和单核巨噬细胞等)浸润心肌组织和大量活性氧(ROS)的释放,同时引起多种促炎细胞因子的分泌和释放。心肌梗死后心肌细胞的坏死激发了炎症反应,炎症反应参与了再灌注损伤的扩展以及梗死后心室重塑的病理生理过程[8]。TNF、IL-1β、IL-6等炎症因子在心肌梗死中显著增加[9-10]。这些炎症因子通过协同作用致血管微循环障碍、血小板聚集及心肌细胞凋亡,增加缺血再灌注损伤。因此,现在越来越趋向于关注炎症反应与心肌IRI之间的紧密联系。
2 IL-17及其受体的信号转导通路
IL-17细胞因子家族包括IL-17A、IL-17B、IL-17C、IL-17D、IL-17E和IL-17F。IL-17A与IL-17F是最主要的促炎细胞因子,它们在氨基酸序列上同源性高达55%,并且两者可以以同源二聚体或异源二聚体的形式存在[11-12]。IL-17A主要由辅助性T细胞17(T helper cell 17,Th17)分泌,另外,一些天然免疫细胞如细胞毒性CD8+T细胞、γδT细胞、自然杀伤T细胞(natural killer T cell,NKT)和B细胞在天然免疫反应早期也可以表达IL-17A。IL-17受体是一种异聚体复合物,由IL-17RA、IL-17RB、IL-17RC、IL-17RD和IL-17RE组成[13-14]。IL-17受体含有保守的结构基序,包括一个细胞外纤维凝集素Ⅲ样结构域和一个细胞质SEFIR结构域。IL-17A和IL-17F形成同二聚体或异二聚体以结合IL-17RA和IL-17RC异二聚体复合物,从而激活下游细胞内信号传导途径,包括核因子-κB(NF-κB),CCAAT/增强子结合蛋白(C/EBPs)、C/EBPβ和C/EBPδ、丝裂原活化蛋白激酶(MAPKs)、JAK-PI3K和JAK-STAT通路。IL-17通过调节粒细胞集落刺激因子(G-CSF)及其受体的表达而扩增中性粒细胞,并且调节趋化因子的表达而募集中性粒细胞[15]。IL-17还能上调组织细胞上细胞间黏附分子-1(ICAM-1)的表达,介导炎性细胞和T细胞在组织中的浸润,并与IL-1β、TNF-α、IFN-γ等细胞因子发挥协同效应,促进T细胞的活化并协助B细胞产生抗体,放大靶器官的免疫反应及炎性破坏从而参与多种感染、慢性炎性疾病、自身免疫性疾病的发生与发展[16]。
3 IL-17在心肌IRI中的作用
局部炎症、细胞凋亡和自噬都是心肌IRI的原因之一。研究表明,IL-17A与多种心血管疾病的发病机制有关,包括动脉粥样硬化、高血压、病毒性心肌炎和扩张型心肌病[17-22]。最近,有研究发现T淋巴细胞介导的免疫炎症反应在IRI中起重要作用[23]。心肌再灌注诱导T细胞活化,活化的STAT3可增强IL-17的表达。Barry等[24]发现,IL-17A和IL-17A受体在心肌IRI后的表达增强。此后,越来越多的证据表明,IL-17A通过促进心肌细胞凋亡、中性粒细胞浸润、促进心肌重塑参与了心肌IRI的发生。IL-17作为一种特殊的促炎细胞因子,对不同类型的细胞具有强大的作用,在心肌IRI期间连接着内源性免疫和适应性免疫[25]。IL-17在再灌注损伤中的机制可能是通过Stat3-iNOS途径、MAPK和PI3K依赖性、HMGB1-IL-23/IL-17途径、HMGB1-TLR4-IL-17A途径,增加IL-17靶基因Cxcl1、Cxcl2、IL-1β、iNOS和IL-6等的表达实现的。IL-17诱导Cxcl-1和IL-6的产生和释放,Cxcl-1是一种重要的趋化因子,它能使中性粒细胞浸润到梗死部位。IL-6等炎性细胞因子和中性粒细胞的聚集打破了促凋亡和抗凋亡蛋白表达之间的平衡,最终触发凋亡程序[26]。Cxcl-1和IL-6的过度表达是由MAPK-PI(3)K/AKT通路激活介导的。Barry等[24]研究结果表明IL-17介导的Cxcl-1表达与ERK1/2、p38或PI(3)K的抑制有关,而激活的JNK可以抑制IL-17依赖的IL-6表达,见图1。
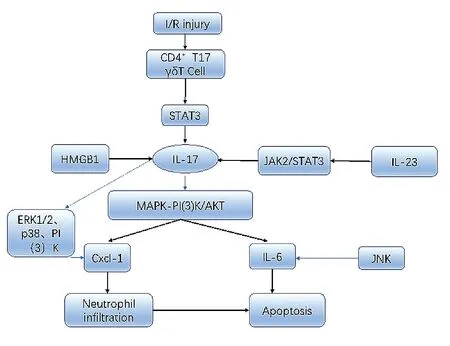
图1 IL-17在I/R损伤中的作用
廖芫熙[27]研究发现在大鼠心肌缺血再灌注模型中,再灌注4 h后心肌组织中IL-23、IL-17A表达增加,给予外源性IL-23处理可以增加心肌梗死面积、心肌细胞凋亡水平、心肌损伤标志物水平以及IL-17A、TNF-α和IL-6等炎症因子的表达,同时I/R诱导的氧化应激水平增加;而中和IL-23能明显减轻心肌IRI。进一步研究发现,在心肌IRI中IL-23可以激活JAK2/STAT3信号通路,而抑制该信号通路可以下调IL-23对IL-17A的调节效应,同时减轻IL-23促炎和促细胞凋亡等加重心肌损伤的效应。以上研究结果提示,在心肌IRI中IL-23可能通过激活JAK2/STAT3信号通路上调IL-17A表达而发挥了加重心肌损伤的效应。
心肌IRI后梗死区边缘存在心肌细胞凋亡,且在急性心肌IRI中发挥着重要作用[28-29]。据报道,IL-17A诱导心肌细胞凋亡并促进心肌梗死后心室重构[30]。此外,IL-17A在心脏移植模型中显著增加,可诱导心肌细胞凋亡,促进促炎介质的释放,在心肌IRI中起重要作用[31-32]。郭梅等[33]研究发现,小鼠心肌缺血再灌注后3、24、72 h时间点心肌组织IL-17A表达显著增加,心肌细胞凋亡率和Caspase-3活性亦明显增加;细胞水平研究发现,不同浓度IL-17A能够促进原代心肌细胞凋亡,诱导Caspase-3、Bax表达,抑制Bcl-2及PI3K/Akt通路蛋白表达。证实心肌IRI小鼠IL-17A明显增加,高表达的IL-17A可能通过抑制PI3K/Akt通路来诱导心肌细胞凋亡。
自噬在心肌IRI的过程发挥了重要作用[34-35]。有研究表明,高迁移率族蛋白1(HMGB1)对自噬有调控作用[36],IL-17A也与自噬有密切关系[37]。许卫攀[38]在心肌细胞缺氧/复氧(H/R)模型中发现,用中和抗体下调HMGB1和IL-17A表达量后,H/R心肌细胞的损伤、氧化应激水平和凋亡率明显降低,自噬相关蛋白LC3和Beclin-1蛋白表达量也相应降低,而给予外源性HMGB1和IL-17A因子后心肌细胞损伤、氧化应激水平和凋亡率明显升高,LC3和Beclin-1蛋白表达量也相应提高,提示HMGB1和IL-17A可以调控心肌细胞H/R时的自噬水平,提高氧化应激水平,促进凋亡,加重心肌细胞损伤。Liao等[31]发现IL-17A介导心肌细胞凋亡,调节Bax/Bcl-2比例,诱导CXC介导的趋化因子中性粒细胞迁移,并诱导内皮细胞E-选择蛋白和细胞间黏附分子的中性粒细胞-内皮细胞黏附1表达。
综合以上研究结果,IL-17在心肌IRI中起重要作用,而IRI发生在再灌注最初的几分钟内,那么IL-17A在缺血再灌注中的表达曲线是怎样的呢?Liao等[31]研究了再灌注后不同时间点的IL-17A的水平。结果发现,IL-17A mRNA和蛋白的表达在再灌注后1 h即显著增加,逐渐上升至24 h时达到高峰,与假手术组相比,在再灌注后72 h仍处于较高水平。Hashmi等[39]发现,稳定型和不稳定型冠心病的患者中IL-17A显著增加,但在再灌注过程中其峰值尚不明确。
在再灌注治疗的患者中,IL-17A水平与心肌梗死面积之间的关系仍存在争议。Cheng等[18]研究表明,与不稳定型心绞痛患者相比,IL-17A在心肌梗死患者中明显升高。Bochaton等[40]首次探讨了在ST段抬高型心肌梗死患者中,IL-17A在再灌注最初数小时的水平和活性以及与其他细胞因子、梗死面积之间的关系。结果表明,与健康对照组相比,心肌梗死患者在再灌注前和再灌注后的最初数小时中IL-17A显著增加。无论是通过肌钙蛋白检测还是心脏磁共振检查,IL-17A水平和活性在心肌梗死急性期与梗死面积均无关。另外,心肌梗死患者的血清可以诱导内皮细胞分泌IL-8,而IL-8是一种强大的白细胞趋化因子。因此IL-17可能通过激活内皮细胞参与中性粒细胞的募集。IL-17A在心肌梗死后的最初几小时轻度激活,可能在后期阶段才显著升高,因此,在人类中IL-17A可能参与了再灌注后期的炎症反应。
4 IL-17家族的药物作用靶点及展望
随着再灌注治疗在临床中越来越广泛的应用,现代医学的治疗重点已逐渐从减少缺血性损伤转向减少再灌注损伤。因此研究IRI的机制并寻找减轻再灌注损伤的合适靶点具有重要的临床意义。作为促炎细胞因子,IL-17促进心肌IRI的发生及发展,提示通过相关药物治疗抑制IL-17炎性反应轴可以减轻心肌IRI。Zhang等[41]发现,在小鼠心脏移植的IRI模型中,Nec-1降低了心肌细胞坏死和中性粒细胞和巨噬细胞的募集。Nec-1可减少心肌IRI后24 h肌钙蛋白T(TnT)的产生、改善心功能、延长心脏移植物存活时间,并且可以减少ROS的产生,增加NOS2和COX-2的表达。Nec-1可降低HMGB1、IL-23和IL-17A的表达。此外,外源性HMGB1可阻断Nec-1诱导的TnT表达降低。总之,Nec-1在心肌细胞IRI中起保护作用,这与抑制HMGB1-IL-23/IL-17通路有关。
综上所述,IL-17通过不同的信号通路参与了心肌缺血再灌注损伤的发生,对IL-17作用信号通路的研究,将为减轻心肌IRI寻找有效的药物作用靶点提供理论依据,因此,这方面的研究可能是未来科研及临床工作的新重点。
