Transcriptome prof ile of Dunaliella salina in Yuncheng Salt Lake reveals salt-stress-related genes under diff erent salinity stresses*
2021-12-09FanGAOFangruNANJiaFENGJunpingQiLIUXudongLIUShulianXIE
Fan GAO, Fangru NAN, Jia FENG, Junping LÜ, Qi LIU, Xudong LIU, Shulian XIE
School of Life Science, Shanxi Key Laboratory for Research and Development of Regional Plants, Shanxi University, Taiyuan 030006, China
Abstract Salt stress is an abiotic stress to plants in especially saline lakes. Dunaliella, a halophilic microalga distributed throughout salt lakes and seas, can respond to diff erent salinity stresses by regulating the expression of some genes. However, these genes and their function and biological processes involved remain unclear. Prof iling these salt-stress-related genes in a high-salt-tolerant Dunaliella species will help clarify the salt tolerance machinery of Dunaliella. Three D. salina_YC salt-stress groups were tested under low (0.51 mol/L), moderate (1.03 mol/L), and high (3.42 mol/L) NaCl concentrations and one control group under very low (0.05 mol/L) NaCl concentration and 3 transcriptome results that were deep sequenced and de novo assembled were obtained per group. Twelve high-quality RNA-seq libraries with 46 585 upregulated and 47 805 downregulated unigenes were found. Relative to the control, 188 common diff erentially expressed genes (DEGs) were screened and divided into four clusters in expression pattern.Fifteen of them annotated in the signif icant enriched Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were validated via qPCR. Their qPCR-based relative expression patterns were similar to their RNA-seq-based patterns. Two signif icant DEGs, the geranylgeranyl diphosphate synthase coding gene (1 876-bp cDNA) and diacylglycerol O-acyltransferase coding gene (2 968-bp cDNA), were cloned and analyzed in silico. The total lipid content, superoxide dismutase specif ic activity, and betacarotene content of D. salina_YC increased gradually with increasing salinity. In addition, the expression of 11 validated genes involved in fatty acid biosynthesis/degradation, active oxygen or carotenoid metabolisms showed signif icant changes. In addition, algal photochemical effi ciency was diminished with increasing salinity, as well as the expression of 4 photosynthesis-related genes. These results could help clarify the molecular mechanisms underlying D. salina responses to the Yuncheng Salt Lake environment and lay a foundation for further utilization of this algal resource.
Keyword: Dunaliella salina; transcriptome analysis; de novo assembly; salt stress
1 INTRODUCTION
As the primary saltwater lake in Shanxi, China,Yuncheng Salt Lake, with an area of 132 km2(Wang et al., 2014), is well known for its rich mineral resources and unique halophilic algae (approximately 222 species) (Xie et al., 1998). Considering that no living aquatic animals have been found at present in the salt lake, whose salinity ranges 0.002-4.2 mol/L(sodium chloride concentration is def ined as salinity in this study) (Xie et al., 1998), the lake is commonly known as the “Dead Sea of China”. The nonmodel unicellular green microalgaDunaliellasalina, aDunaliellaspecies that can survive in saline environments, was f irst found in the salt lake in the 1980s (He et al., 1993; Xie et al., 1998). Research has demonstrated that this green alga can grow rapidly,accumulate lipids, and expand its cellular capacity under diff erent concentrations of NaCl stress (Cheng et al., 2019). Due to its unique lipid-producing feature,D.salinamay represent a potential new target for biofuel feedstock (Ahmed et al., 2017). Moreover,this green microalga has been widely exploited in the medical and food industries due to its unique intracellular metabolite beta-carotene, which can be enriched through the regulation of algal culture conditions (BenMoussa-Dahmen et al., 2016).
NaCl not only constitutes a necessary nutrient for the normal growth ofD.salinabut also serves as a natural and complex abiotic stress factor inf luencing the expression of genes involved in various biological processes including metabolic, physical, and chemical processes. We postulated that diverse salt-stressrelated genes and the biological processes in which they are involved might be responsible for the abovedescribed features ofDunaliella. By reviewing numerous studies, we found that some essential metabolic pathways, for example, photosynthesis(Wei et al., 2017), lipid metabolism (Azachi et al.,2002) and carotenoid metabolism (Wichuk et al.,2014; Solovchenko et al., 2015), might be inf luenced by salt tolerance inDunaliellacells. The key factors inf luencing these metabolic processes primarily include salt stress genes (Li et al., 2010), halotolerant proteins (Wang et al., 2019), and metabolic enzymes(Wu et al., 2019). The genes encoding photosystem II protein D1 (PsbA) in photosystem II (PSII)(Vasilikiotis and Melis, 1994) and photosystem I P700 chlorophyllaapoprotein A2 (PsaB) in photosystem I (PSI) (Liang et al., 2008) ofD.salinahave been cloned successfully, and the former’s expression has been conf irmed negatively correlated with the environmental salt concentration of this green microalga. Lipid metabolism-related enzymes(Davidi et al., 2012), such as triglyceride lipase,glycerol kinase, and glycerol dehydrogenase, have been proposed to be involved in controlling diff erent types of resistance ofD.salinato salinity stress through self-regulation of algal cellular osmotic pressure. As an enzyme involved in carotenoid metabolism, lycopene β-cyclase was reported to accumulate inDunaliellasp. in a high-salinity water environment, with the expression of its coding gene(LCYb) attributed to the salt-responsive element GT-1 motif in soybean CaM isoform-4 (GT1GMSCAM4)(Liang et al., 2017). Similarly, specif ic antioxidant enzymes and stress proteins involved in environmental adaptation, such as peroxiredoxins (Gong et al., 2017)and heat shock protein 90 (Chen et al., 2015), have been shown positively correlated with the response ofD.salinato salt stress. However, the genes encoding many key metabolic enzymes involved in the response to salt stress have not yet been characterized inD.salina. Thus, novel salt-stress-related genes may be discovered based on transcript data fromD.salinaunder diff erent salinity stresses.
Dunaliellais facing a potential survival crisis due to habitat threats, such as environmental pollution,which has been increased in Yuncheng Salt Lake and salt lakes in other areas in recent years (Yuan et al.,2015). To date, only 143 known functional genes have been reported in GenBank (July 2020). To explore new and more genes, particularly those essential for the regulation of salt tolerance inD.salina, transcriptomic prof iling of this unique microalga is urgently required. Due to the rapid development of molecular biology and bioinformatics in the 21stcentury, high-throughput RNA sequencing(RNA-seq) has become an eff ective approach for obtaining transcriptomic information of organisms(Zhou et al., 2019), which is essential for discovering key genes encoding regulators, enzymes or proteins involved in diverse biological processes in algae,such asD.salina. The functions of these genes and their relevant metabolic pathways in this microalga could be further examined using bioinformatic analysis (Rodríguez-García et al., 2017), setting a foundation for research on the regulatory mechanisms of salt tolerance or the survival response ofD.salinain diff erent saline environments. However, such transcriptome analyses with diff erent concentrations of NaCl treatments have not been conducted to date for any wild high-salt-tolerantDunaliellaspecies.Therefore, the key salt-stress related genes and their expression patterns, potential functions, and associated biological processes inD.salinaunder diff erent salinity stresses need to be characterized.
High-throughput sequencing technologies, such as Ion Torrent, Roche 454, Thermo Fisher, SOLiD,HiSeq and, most notably, RNA-seq (Gierahn et al.,2017), have become eff ective approaches for uncovering new functional genes, which may inform the development of further studies on the molecular mechanisms underlying adaptation to abiotic stress in algae. Moreover, algal cell morphology, gene expression, and physiological and biochemical characteristics will be inf luenced by diff erent NaCl concentrations, as reported in various algal species,such asSpirogyrapratensis(Van De Poel et al., 2016),Shewanellaalgae(Fu et al., 2014),Chlorellasp.(Mansfeldt et al., 2016),Pyropiatenera(Im et al.,2017), andChromochloriszof ingiensis(Mao et al.,2020). Furthermore, transcriptomic analysis of a mutantD.tertiolectaunder low-light stress based on the Illumina MiSeq system revealed 10 185 genes and a fatty acid biosynthesis pathway involved in inositol phosphate metabolism (Yao et al., 2015). Subsequent transcriptomic analysis ofD.tertiolectaunder nitrogen def iciency stress using the Illumina MiSeq and HiSeq 4000 systems identif ied 17 845 proteincoding transcripts (Yao et al., 2017). Moreover, some key genes and related metabolic pathways inD.tertiolectaunder nitrogen and NaCl stresses were identif ied via Roche 454 sequencing (Rismani-Yazdi et al., 2011). Finally, comparative transcriptomic analysis ofDunaliellaacidophilaunder cadmium and natural metal-rich stresses using Illumina sequencing revealed specif ic key genes responsive to the heavy metal stress response (Puente-Sánchez et al., 2016).
In addition to transcriptomic studies on otherDunaliellaspecies under diff erent stresses, only two transcriptome sequencing analyses ofD.salinahave been reported to date. For one of the studies, the sequencing platform and abiotic stress conditions imposed duringD.salina’s entire growth cycle were not clarif ied (Hong et al., 2017). For the other, a short stress cycle (0-2 h) and single high-salinity stress(2.5-mol/L NaCl) condition forD.salinaand transcriptome sequencing with the Illumina HiSeq 2000 system were reported (He et al., 2020).Additionally, transcriptome sequencing ofD.salinafrom Yuncheng Salt Lake has not been performed to date. Hence, numerous unknown salt tolerance- or stress-related genes have yet to be identif ied. Our study sought to perform transcriptome sequencing of the wild halophytic algaD.salina, which can tolerate high salinity, collected from the natural salt lake in Yuncheng. To identify the regulated or responsive key genes that may be associated with the salt tolerance ofD.salinain this environment, 9 RNA-seq libraries under three diff erent salinity stress conditions were constructed and deep sequenced for this microalga on the new next-generation sequencing platform BGISEQ-500, which has been successfully used in the RNA-seq of many species (Mak et al., 2017).Based on the results of de novo transcriptome sequencing, morphological, physiological, and biochemical analyses of this alga, we characterized its salt-stress-related genes, screened for enriched diff erentially expressed genes (DEGs), predicted the functions and related biological processes of the DEGs, and conducted gene expression validation and molecular cloning, which will prove helpful for further demonstrating the salt tolerance or response mechanism of this alga. Key gene information will provide insight for improving the quality ofD.salinaat the molecular level.
2 MATERIAL AND METHOD
2.1 Collection and cultivation of microalgal samples
Samples ofD.salina(referred to here asD.salina_YC) were collected from Yuncheng Salt Lake in Shanxi, China (approximately 110°10′30″E ,35°1′10″N), with an environment salinity of~3.66-mol/L sodium chloride as detected by a BSD-200 salinometer (BELL Analytical Instruments, Co.,Ltd., Dalian, Liaoning, China). Following isolation of one pureD.salina_YC strain from these wild samples using capillary and microscopy techniques, the strain was f irst identif ied and cultured on BG11 medium with 0.05-mol/L NaCl, 0.017-mol/L NaNO3,2.0×10-4-mol/L K2HPO4, 3.0×10-4-mol/L MgSO4·7H2O,2.0×10-4-mol/L CaCl2·7H2O, 2.0×10-4-mol/L Na2CO3,3.12-mol/L C6H8O7, 2.14-mol/L C6H10FeNO8, and ultrapure water added to 2.0 L in a pH 7.2 environment.Next, we extracted 1.5-L fresh liquid algae cultured under 10 000-lx irradiance provided by day-light f luorescent tubes for 12 h/d at 20 °C in an indoor algal cultivation room. Finally, 150-mL liquid algal solutions from the same strain were exposed to four diff erent salinities (0.05-, 0.51-, 1.03-, and 3.42-mol/L NaCl) by adding the corresponding weight of sodium chloride to the medium. We named these groups as DS_CO, DS_LS, DS_MS, and DS_HS, respectively.DS_CO cultured in raw BG11 medium with 0.05-mol/L NaCl was used as the control group in this study. Moreover, to ensure normal algal growth and avoid microbiological contamination, theD.salina_YC cultures were microscopically examined every 8 d, with necessary nutritional supplements added.The cell densities of DS_CO, DS_LS, DS_MS, and DS_HS during diff erent growth stages were calculated by measuring the optical density (OD) value of liquid algae at 685 nm with a signif icant optical absorption peak on a UV2600 system (Shimadzu Co., Ltd.,Shanghai, China) and drawing a standard curve between cell density and corresponding OD. A f lowchart of the entire experiment forD.salina_YC in this study is shown in Supplementary Fig.S1.
2.2 RNA isolation, cDNA library construction and transcriptome sequencing
To ensure the accuracy of sequencing, three biosample replicates of DS_CO, DS_LS, DS_MS,and DS_HS from the above algal strain on the 51st day were selected for RNA isolation (Supplementary Fig.S1). Each sample was enriched by centrifugation(2 500×g) at 4 °C and quick-frozen in liquid nitrogen.Next, the pools of total RNA ofD.salina_YC were extracted and purif ied using TRIzol reagent(Invitrogen Life Technologies Inc., Carlsbad, CA,USA). The quality and quantity of the extracted total RNA were estimated using an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA).Subsequently, paired-end cDNA libraries were constructed using a SMART cDNA Library Construction Kit (TaKaRa, China) according to the manufacturer’s instructions. Sequencing of the transcriptome was conducted using the BGISEQ-500 platform developed in 2015 by Beijing Genomics Institute (BGI), a new convenient high-throughput sequencing platform combined with probe-anchored polymerization and improved DNA nanosphere technology (Mak et al., 2017). Raw sequencing data were deposited in NCBI Short Read Archive (SRA) in 2019 and 2020. The SRA accession numbers can be seen in Data Availability Statement.
2.3 Sequence analysis and de novo transcriptome assembly
The preliminary transcriptomic library data were f iltered and checked using SOAPnuke (https://github.com/BGI-f lexlab/SOAPnuke) (Chen et al., 2018).Brief ly, reads containing adapters and poly-N (N>5%)on the 5′ and 3′ ends were removed, and low-quality reads were trimmed. Next, the clean reads meeting quality 20 (Q20) and quality 30 (Q30) criteria(Q20>95%, Q30>85%) were screened. Further, the lengths of all unigenes containing clusters and singletons were obtained using TGICL (Pertea et al.,2003). Finally, the quality of theD.salina_YC transcriptome assembly was evaluated with BUSCO software (https://busco.ezlab.org/) using default parameters (Seppey et al., 2019).
2.4 Annotation of the transcriptome
Based on the best sequence similarity alignment(E-value≤1 E-10, length percentage of the query sequence≥85%, and nonredundant contigs), functional annotation of the transcriptome was obtained by comparing the assemblies against the Gene Ontology(GO), RefSeq nonredundant protein (NR), nucleotide sequence (NT), Swiss-Prot, Pfam, Kyoto Encyclopedia of Genes and Genomes (KEGG), and euKaryotic Orthologous Group (KOG) databases. GO terms were described using Blast2GO v2.5.0 (Götz et al., 2008).Unigenes assigned a corresponding protein function were annotated in the Swiss-Prot, Pfam, and KEGG pathways with diff erent KEGG orthologies (KOs).All unigenes were aligned to the KOG database and given corresponding protein IDs to obtain annotation results. Categorized information for the screened unigenes with coding sequence (CDS) regions for protein synthesis in eukaryotes (including plants and fungi), bacteria, viruses, and protozoans were predicted according to the NR database (Han et al.,2018). Moreover, poential transcription factors (TFs)were predicted by aligning the open reading frames(ORFs) of unigenes against the sequences stored in the Pfam and PlantTFDB databases with the HMMER program (Jin et al., 2017). All output results for gene annotation were further processed and cleaned to remove duplicates and select the best-matching annotation.
2.5 Gene expression analyses
Potential genes were obtained through the alignment of clean reads for eachD.salina_YC sample against the corresponding de novo transcriptome reference using Bowtie2 software(http://bowtie-bio.sourceforge.net/Bowtie2/index.shtml)(Langdon, 2015). Next, the gene expression for each algal sample based on RNA-seq was calculated using fragments per kilobase of transcript per million fragments mapped (FPKM) data (Tian et al., 2019).All samples were clustered via principal component analysis (PCA) based on their global gene expression levels. Subsequently, the DEGs for each sample were calculated using the RSEM v1.2.12 package with default parameters (http://deweylab.biostat.wisc.edu/rsem/rsem-calculate-expression.html) (Djebali et al.,2017). The number and distribution of DEGs in the algal samples were screened using DESeq2 software with a fold change ≥|2.00| and adjusted p-value (false discovery rate, FDR) ≤0.05 (Maza, 2016), while signif icant DEGs were screened with a fold change>|32| and FDR<0.03. Furthermore, a clustering heat map of DEGs from the three algal comparison groups, the salt-stress groups relative to the control group (DS_LS vs. DS_CO, DS_MS vs. DS_CO, and DS_HS vs. DS_CO) with diff erent FPKM values was constructed by running the heatmap function in R (Hu et al., 2019). Similarly, the expression patterns of DEGs with similar expression trends were predicted by running Mfuzz v2.34.0 (http://mfuzz.sysbiolab.eu)(Kumar and Futschik, 2007).
2.6 Enrichment of GO and KEGG pathways
Blast2GO software was used to determine the enriched GO terms of the common DEGs among the diff erentD.salina_YC experimental comparison groups and further categorize them into 3 GO functional groups, biological process (BP), molecular function (MF), or cellular component (CC), withQ-values less than 0.01 (Miura et al., 2019). Similarly,the common DEG data-assigned KO IDs were submitted to the KEGG Automatic Annotation Server(KAAS) and mapped to KEGG pathways categorized into diff erent KEGG functional groups, including cellular process, environmental information processing,genetic information processing, metabolism, and organismal system (Moriya et al., 2007). The KEGG pathway enrichment results were further analyzed by using the phyper function of R software with aQ-value less than 0.01 (Lee et al., 2019).
2.7 Quantitative PCR
Fifteen common signif icantly enriched DEGs among the 3D.salina_YC experimental groups as detected by GO and KEGG pathway analyses were selected for real-time quantitative PCR (qPCR)validation in this study. Total RNA was extracted from each experimental group using the RNAsimple Total RNA Kit (DP419) according to the manufacturer’s instructions (Tiangen, China). Next, total RNA was used to synthesize cDNA using the PrimeScriptTMRT reagent kit according to the manufacturer’s instructions(TaKaRa, China). Subsequently, each qPCR was performed in a f inal 20-μL reaction volume containing 2-μL cDNA template, 8-μL SYBR Premix Ex Taq(TaKaRa), 2-μL primers at a 2.5-μmol/L concentration,and 8-μL distilled water. Primers were designed based on the transcripts and synthesized by Invitrogen Biotech Co., Ltd. (Shanghai, China). Finally, qPCR was performed using the Applied Biosystems StepOnePlusTMReal-Time System (Thermo Scientif ic,Waltham, MA, USA) with the following cycle conditions: 95 °C for 1 min, followed by 40 cycles of 95 °C for 15 s, 50-58 °C for 20 s (depending on the optimal melting temperature for each primer pair), and 72 °C for 20 s, with a f inal 72 °C step for 5 min.

2.8 Cloning and sequence analysis of GGPS and DGAT from D. salina_YC
The average fold changes of the common DEGs based on RNA-seq and qPCR data showed thatggps(unigene 47314, geranylgeranyl diphosphate synthase gene) anddgat(unigene 16111, diacylglycerol O-acyltransferase gene) were the relatively signif icant.We f irst selected them for cDNA full-length cloning inD.salina_YC. Based on the two genes’ transcripts,we designed corresponding nested primer pairs to clone the full-length cDNAs using a SMART RACE Kit (TaKaRa). According to the manufacturer’s instructions, the PCR systems were prepared for a minimum of three PCR rounds with the cDNA ofD.salina_YC samples mixture under 4 salinity conditions as the template. The f irst PCR was conducted with the following program: 95 °C for 3 min, followed by 5 cycles of 94 °C for 30 s and 72 °C for 3 min; another 5 cycles of 94 °C for 30 s,70 °C for 30 s, and 72 °C for 3 min; a f inal 20 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 3 min;and f inally, 72 °C for 5 min. Next, nested PCR was conducted with the following PCR program: 94 °C for 3 min, followed by 25 cycles of 94 °C for 30 s,55 °C for 30 s, and 72 °C for 3 min, and f inally, 72 °C for 5 min. The cDNA sequences and amino acids of the genes were analyzed using the following bioinformatic tools: BLASTn and BLASTp for sequences alignment analysis, ORFf inder (https://www.ncbi.nlm.nih.gov/orffi nder/) for ORF detection(Wheeler et al., 2004), DNAMAN v.4.0 for prediction of amino acids (Song et al., 2009), ExPASy (http://cn.expasy.org) for prediction of protein pI and calculation of molecular weight (MW) (Gasteiger et al., 2003), ProtScale (https://web.expasy.org/protscale/) for detection of protein hydrophobicity(Seddigh and Darabi, 2018), SWISS-MODEL(https://www.swissmodel.expasy.org/) for prediction of protein 3D model structure (Biasini et al., 2014),Clustal v.2.0 for multiple amino acid sequence alignment, G-blocks for redundant data removal, and MEGA v.6.0 software (Jin et al., 2015) for construction of a protein phylogenetic tree with the maximumlikelihood (ML) algorithm. The GTR substitution model was used for ML tree construction, with a discrete gamma distribution among amino acid sites and a gamma-distributed with invariant sites (G+I)rate among sites. The bootstrap consensus ML tree was inferred from 1 000 replicates.
2.9 Analysis of physiological and biochemical characteristics
The total lipid content (TLC) (mg/cell) of eachD.salina_YC experimental group at diff erent growth periods (1, 6, 11, 16, 21, 26, 31, 36, 41, 46, and 51 d)was measured using the ratio of total lipids detected with the chloroform-methanol method to algal cell quantity (Huang and Kim, 2017). The photochemical(PC) effi ciency of PSII was determined for the 4D.salina_YC experimental groups during the same growth periods above by measuring the ratios of variable f luorescence (Fv)/maximum f luorescence(Fm) andFv/minimum f lorescence (Fo) (Wang et al.,2017) on a CCM-300 chlorophyll f luorescence monitor (Opti-Sciences Inc., Boston, OH, USA). The superoxide dismutase (SOD) specif ic activity (SSA)(U/mg) of eachD.salina_YC experimental group at 51 d at diff erent stress times (1, 10, 20, 30, 40, 50, and 60 min) was detected by calculating the ratio of total SOD activity (Zhang et al., 2014) detected with a SOD Assay Kit (BioVision Inc., San Francisco, CA,USA) to algal total protein content detected with the trichloroacetic acid-acetone precipitation method(Wu et al., 2014). The beta-carotene content (BCC)(mg/cell) of eachD.salina_YC experimental group at diff erent growth phases was evaluated by calculating the ratio of beta-carotene quality to algal cell quantity.Beta-carotene quality was calculated by detecting the OD values of algal beta-carotene extracts at 450 nm(a prominent peak appears at this wavelength)combined with the beta-carotene concentration standard curve ofD.salina_YC (Fazeli et al., 2006).All physiological and biochemical indexes for theD.salina_YC experimental groups were calculated with three replicates for each sample. Student’st-tests were used for statistical analyses. Signif icance was annotated using diff erent types of letters or diff erent numbers of asterisks.
3 RESULT
3.1 Morphology and growth characteristics
Morphological observation combined with growth kinetics of theD.salina_YC cells showed that ~51 d was the stationary phase (Fig.1a & b). Accordingly,this was the optimum time for microscopic observation of the algae, as well as for RNA extraction and sequencing (Table 1). Markedly smaller spherical algal cells (2-4 μm) with lighter pigmentation were observed in DS_CO during the growth stages (mean cell density, 5.337×104cells/mL) compared to the other experimental groups (mean cell density,9.350×105cells/mL) (Supplementary Table S1a),which indicated that NaCl is a necessary nutrient for algal cell development and that a very low concentration of NaCl in the medium was insuffi cient for substantialD.salina_YC indoor growth. Unlike the DS_HS cells with more yellow or yellow-orange pigment (9-13-μm size), which had more lipids and larger cells and pyrenoids, DS_LS and DS_MS cells(3-10-μm size) contained green or yellow-green pigmented cells, fewer lipids, and smaller pyrenoids.In addition, the majority ofD.salina_YC cells had two f lagella, which facilitated their movement and capture of nutrients in the salt water; however, no noticeable eyespots were observed (Fig.1a; Table 1).Furthermore, the order of maximum cell density during the algal stationary phase was DS_LS(2.805×106cells/mL)>DS_MS (2.102×106cells/mL)>DS_HS (8.527×105cells/mL)>DS_CO(1.334×105cells/mL) (Fig.1b, Supplementary Table S1), indicating that low and moderate concentrations of NaCl were benef icial for indoor culture ofD.salina_YC.
3.2 RNA-seq and de novo assembly
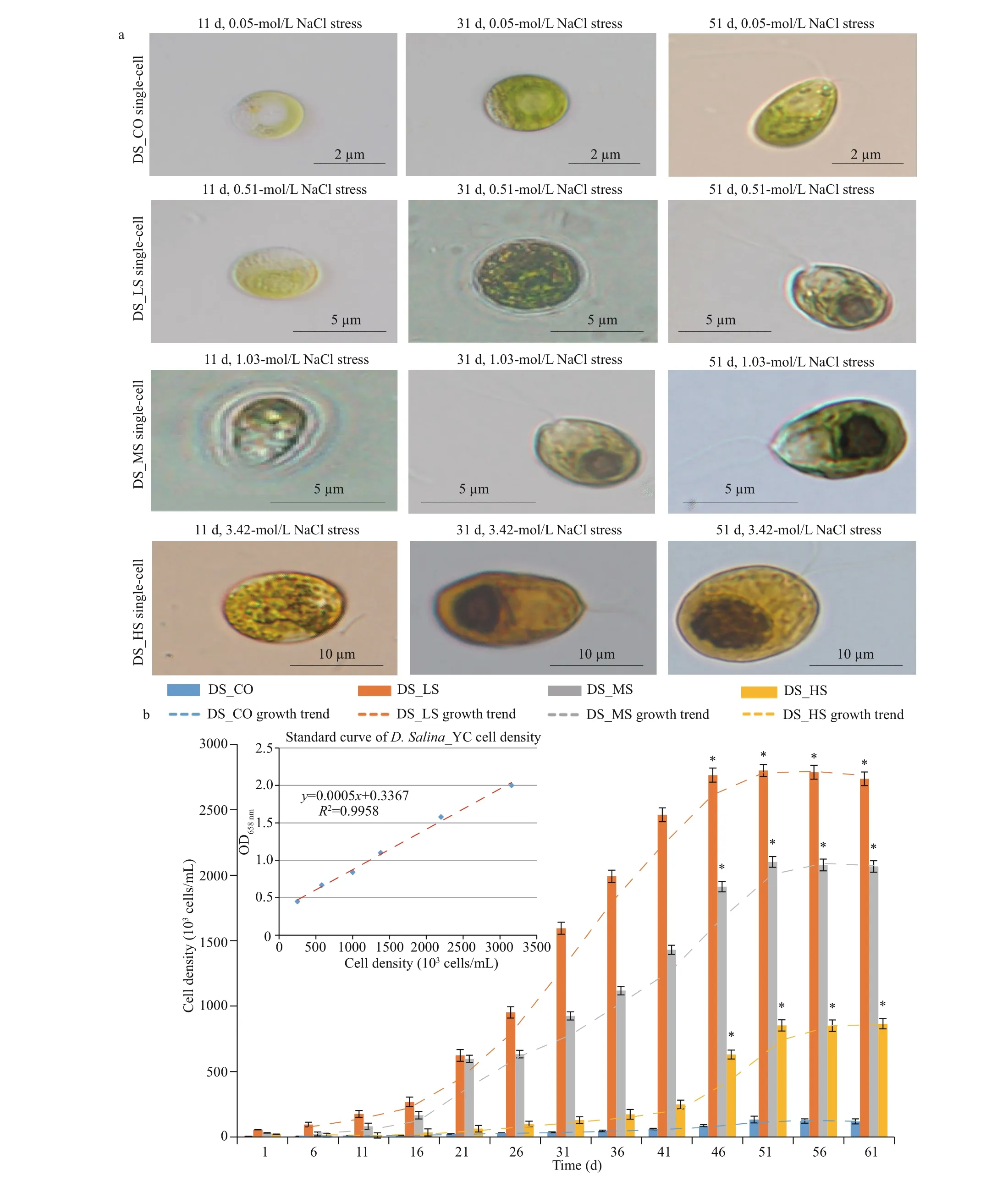
Fig.1 Morphology and growth of D. salina_YC cells under 4 diff erent salinity treatments

Table 1 Morphology and RNA sample information of D. salina_YC at the stationary phase
Although genome annotations ofD.salinawere reported in 2017 (Polle et al., 2017), they were not suitable for the transcriptomic analysis performed in this study, as considerably few eff ective gene annotations (merely 143 eff ective functional genes inD.salina) and a low RNA-seq data percentage(merely approximately 44.06%) mapped to the reference genome Dsal_v1.0 (NSFN00000000.1).Thus, the de novo assembly approach was applied for the YunchengD.salinatranscriptomic analysis in this study.
A total of 886.64 M raw reads was obtained through RNA-seq based on the 12 cDNA libraries for the 4D.salina_YC experimental samples (Supplementary Table S2). An average of 68.64 M clean reads per algal sample (accounting for 92.36% of the total reads) was obtained, with an average contig length of 856 bp after f iltering adapters, sequences with unknown bases (N>5%), and low-quality reads (Table 2). Supplementary Table S2a shows the nucleotide quality of the clean reads (average percentages of Q20 and Q30 were 97.49% and 89.78%). In addition,78.25% of the total clean reads were mapped onto the genome ofD.salina(52.40% of the unique clean reads mapped). The salt-response-related de novo transcriptome ofD.salina_YC was assembled,yielding 116 167 unigenes with a total length of 7.97×107bp (unigene numbers among all 12 algal samples are shown in Supplementary Fig.S2a), and after the removal of redundant data, an N50 of 1 323 bp and a GC percentage of 51.84% wereobtained (Table 2). Completeness of the de novo assembly was investigated using BUSCO assessment,which identif ied 179 complete single copies and 142 complete duplications among all unigenes, yielding an integrity over 90% (Supplementary Fig.S2b). A total of 4 9751 CDSs (total length of 3.96×107bp)were predicted with an N50=978 bp and a GC%=52.95% (Supplementary Table S2b & Fig.S2c).Moreover, 23 595 simple sequence repeat (SSR)markers (total length of 2.285×104bp; Table 2) were detected among 13 621 unigenes with 1-6 nucleotide repeats (Supplementary Table S2c).
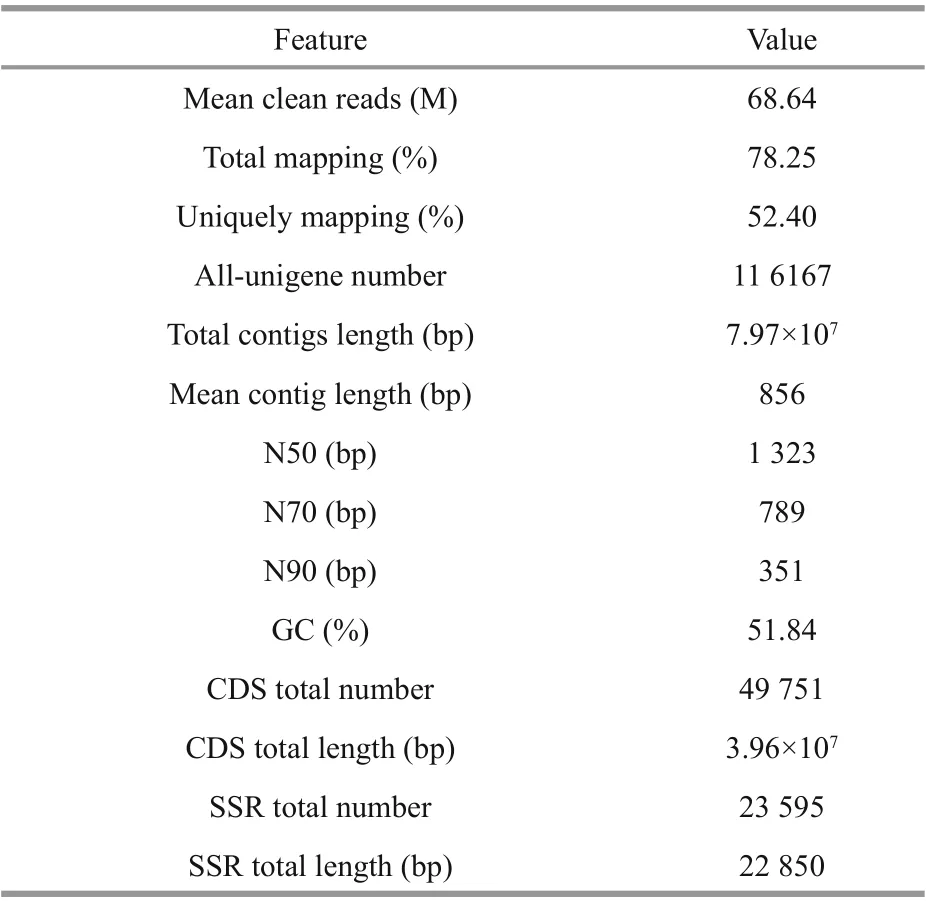
Table 2 D. salina_YC transcriptome assembly details
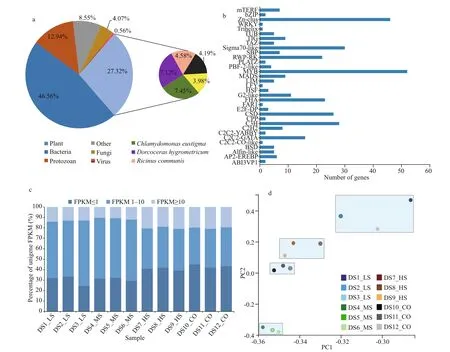
Fig.2 Unigene annotation, expression, and PCA of D. salina_YC under 4 salinity treatments based on transcriptomic data
3.3 Functional annotation and gene expression
Of the 116 167 unigenes (Supplementary Table S3a), 71 932 (79.67%) were annotated using the following databases: NR, NT, Swiss-Prot, KEGG,KOG, Pfam, and GO (Table 3). A total of 4 593(5.05%) unigenes were annotated using all seven databases. Details regarding the quantity of intersecting genes are shown in f ive-dimensional Venn diagrams(Supplementary Fig.S3a-c). Taxonomic analysis demonstrated that 15.62% of the 48 502 screened unigenes with CDS regions were found in three algae,namely,Chlamydomonaseustigma,Goniumpectorale,andVolvoxcarteri, while another 11.70% matched the genes of land plants, namely,DorcocerashygrometricumandRicinuscommunis(Fig.2a;Supplementary Table S3b). In addition, 46.56% of theD.salina_YC unigenes with CDSs were shared with bacteria, 12.94% with protozoans, 0.56% with viruses,and 4.07% with fungi. Last, 4 147 (8.55%) unigenes were shown unique toD.salina_YC (Fig.2a).Moreover, 358 putative genes were predicted in 33 plant TF families, with the highest number of genes(52) predicted in the v-myb avian myeloblastosis viral oncogene homolog (MYB) (Kawase et al., 2017)(Fig.2b).The gene expression densities of the 12 algal samples were diff erent, with two main expression peaks detected (Supplementary Fig.S3d).Furthermore, 8 763 on average of highly expressed unigenes (FPKM≥10) were detected in the 12 algal samples (Fig.2c; Supplementary Table S3c). Most of the remaining unigenes (overall coverage of 87.69%)were expressed at low levels in the algal samples,with an FPKM<10 (Fig.2c; Supplementary Table S3c). Moreover, based on the gene expression information presented above, the three parallel biosamples of eachD.salina_YC experimental or control group clustered together in PCA (Fig.2d;Supplementary Table S3d).
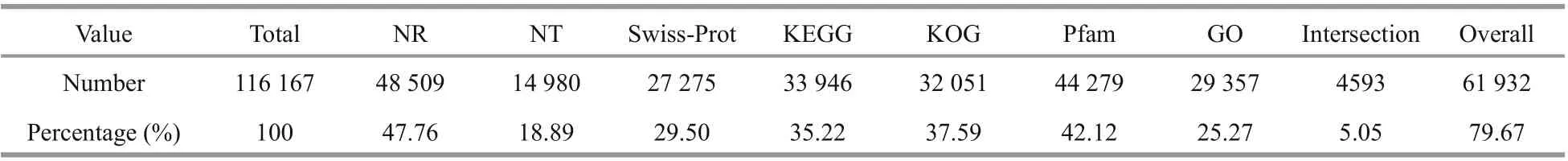
Table 3 Unigene annotation in D. salina_YC
3.4 DEG analysis and clustering
A total of 46 585 upregulated and 47 805 downregulated genes were identif ied without removing the redundant data by comparing the FPKM values of DS_LS to DS_CO, DS_MS to DS_CO, and DS_HS to DS_CO (Supplementary Fig.S4). In addition, some signif icant DEGs (a fold change>|32|,FDR<0.03) were detected by comparing the FPKM values of DS_LS vs. DS_CO (54 upregulated and 41 downregulated DEGs) and DS_MS vs. DS_CO (34 upregulated and 48 downregulated DEGs) (Fig.3a &b; Supplementary Table S4a). Moreover, a few signif icant DEGs could be detected between DS_HS and DS_CO (18 upregulated and 25 downregulated DEGs) (Fig.3c).
In total, 188 common DEGs were obtained by analyzing the intergroup unigenes ofD.salina_YC comparison groups (DS_LS vs. DS_CO, DS_MS vs.DS_CO, and DS_HS vs. DS_CO) (Fig.3d).Additionally, based on the expression levels of these common DEGs, their comparison groups, DS_LS vs.DS_CO and DS_MS vs. DS_CO, grouped together,with more common DEGs upregulated (FPKM≥8.0)than downregulated (Fig.4). More downregulated common DEGs (FPKM≤6.0) were detected in the comparison group of DS_HS vs. DS_CO.Furthermore, we divided them into 4 main clusters(clusters I, II, III, and IV) from 12 cluster blocks based on their relative expression levels (Supplementary Table S4b). Moreover, four diff erent genes relative expression patterns were predicted based on the expression change trend of these genes (Fig.4).
3.5 GO and KEGG analyses of DEGs
The DEGs inD.salina_YC were grouped into diverse functions and pathways according to their assigned GO terms and KO IDs (Supplementary Table S5a & b). The number of DEGs in DS_LS vs. DS_CO was higher than that observed in DS_MS vs. DS_CO and in DS_HS vs. DS_CO (Supplementary Fig.S5a &b). According to the GO classif ication criteria (Han et al., 2018), the majority of the DEGs were assigned to cellular components and biological processes(Supplementary Fig.S5a). Additionally, most of the DEGs were assigned to metabolism pathways according to the KEGG pathway classif ication criteria(Supplementary Fig.S5b) (Lee et al., 2019).
Furthermore, among the top 20 GO enriched terms of the common set of DEGs in the 4 clusters, the terms acetyl-CoA carboxylase activity (Qvalue=1.00E-10, term number=120), photosystem II(Q-value=1.00E-50, term number=3 500), defense response to reactive oxygen species (ROS) and salt stress (Q-value=1.00E-06, term number=28), and carotenoid biosynthesis (Q-value=3.16E-09, term number=2 500) were the most signif icant enriched GO terms in clusters I, II, III, and IV, respectively(Fig.5a-d). Similarly, the top 20 KEGG pathways that were enriched based on the common set of DEGs in the 4 clusters included fatty acid biosynthesis (Qvalue=1.00E-44, pathway number=40),photosynthesis-antenna protein (Q-value=1.00E-75,pathway number=1 500), active oxygen metabolism(Q-value=1.00E-37, pathway number=8), and carotenoid metabolism (Q-value=1.00E-62, pathway number=550) as the most signif icant pathways in clusters I, II, III, and IV, respectively (Fig.6a-d).
3.6 Expression prof iles of common DEGs in the signif icantly enriched GO and KEGG pathways
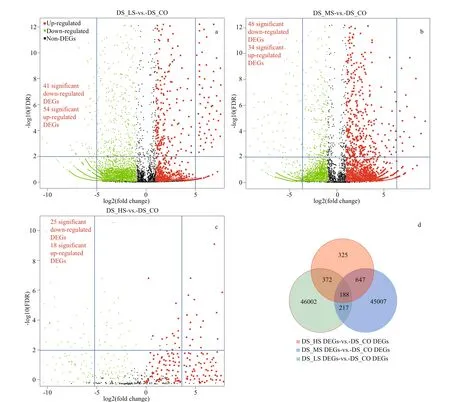
Fig.3 Volcano plot and Venn diagram of diff erentially expressed genes (DEGs) in D. salina_YC under 4 salinity stresses based on transcriptomic data
The expression levels of the common DEGs in the 3D.salina_YC comparison groups involved in the signif icantly enriched biological processes or metabolic pathways were compared via calculating their FPKM expression ratios of salt-stress group to control group (Fig.7a-d). FPKM mean values of the common DEGs in DS_CO classed into cluster I, II,III, and IV were 2.02, 7.23, 2.61, and 1.64, respectively.Downregulated DEGs that globally repressed acetyl-CoA carboxylase activity or fatty acid biosynthesis obtained from GO and KEGG pathway in cluster I were detected in DS_HS vs. DS_CO (FPKM mean ratio of 2.23) relative to DS_LS vs. DS_CO (FPKM mean ratio of 6.26) and DS_MS vs. DS_CO (FPKM mean ratio of 10.28) (Fig.7a). Upregulated expression trends of the DEGs encoding the components of photosystem II or photosynthesis-antenna protein obtained from GO and KEGG pathway in cluster II were overall similar between DS_LS vs. DS_CO(FPKM mean ratio of 15.54) and DS_MS vs. DS_CO(FPKM mean ratio of 16.66) (Fig.7b). Global upregulated DEGs that mediated algal cells defense responses to ROS or salt stresses, or involved in active oxygen metabolism in this alga obtained from GO and KEGG pathway in cluster III were detected in DS_MS vs. DS_CO (FPKM mean ratio of 10.26)and DS_HS vs. DS_CO (FPKM mean ratio of 13.74)(Fig.7c). Overall expression trend of DEGs involved in carotenoid biosynthesis or metabolism obtained from GO and KEGG pathway in cluster IV were upregulated in DS_HS vs. DS_CO (FPKM mean ratio of 7.67) relative to DS LS vs. DS_CO (FPKM mean ratio of 4.98) and DS_MS vs. DS_CO (FPKM mean ratio of 2.88) (Fig.7d).

Fig.4 Heat map and potential expression pattern of the common DEGs among the 3 comparison groups: DS_LS vs. DS_CO,DS_MS vs. DS_CO, and DS_HS vs. DS_CO
3.7 qPCR validation
Fifteen genes were randomly selected from the common DEGs in the most signif icant enriched GO terms and KEGG pathways for qPCR validation.Detailed gene annotation, potential function, and PCR primer information are shown in Supplementary Table S6a & b. The qPCR results showed that all genes were diff erentially expressed among the 4D.salina_YC experimental groups. The diff erential expression trend detected by qPCR was relative identical to that identif ied by RNA-seq (Fig.8a-d; Table 4). Relative to the DEGs of DS_CO, the signif icant diff erential expression oflacs,nadb,dagt,lhcb5,lhcbm8,cpld35,andggpsin otherD.salina_YC experimental groups with higher salinity stress (more than 0.05-mol/L NaCl) could be clearly observed not only in RNA-seq,but also in qPCR analysis (Fig.8a-d). Among these genes,fsd1obtained from the enriched common DEGs in cluster III was markedly upregulated between DS_LS and DS_HS relative to DS_CO based on qPCR analysis (Δ fold change=5.7,P-value<0.01) (Fig.8c).Four genes obtained from clusters I and II, namely,lacs,nadb,lhcb5, andlhcbm8, were upregulated between DS_LS and DS_MS relative to DS_CO based on qPCR analysis (Δ fold change>0.3,P-value<0.05)(Fig.7a & b). Additionally, among these signif icant genes (fold change>10), theggpsobtained from cluster VI was a markedly linearly downregulated gene among the 3D.salina_YC comparison groups with increasing salinity relative to DS_CO (Δ fold change<-1.2,P-value<0.01) (Fig.8d). Furthermore,the genedgatobtained from cluster I markedly upregulated between DS_LS and DS_MS relative to DS_CO (Δ fold change=3.3,P-value<0.001), but markedly downregulated between DS_LS and DS_HS relative to DS_CO (Δ fold change=-24.2,P-value<0.001) (Fig.8a).

Table 4 15 common DEGs annotation information and their relative expression levels relative to the control (DS_CO)
As shown in Table 4, the functions of the 4 validated genes,lacs,nadb,dgat, andacat1, are related to some key enzymes regulating the processes of fatty acid biosynthesis and degradation. Among the genes,dgatwas the best candidate gene for its full cDNA clone based on its relative signif icant expression change in the 3 comparison groups (Pvalue at least less than 0.01) measured by qPCR(Table 4; Fig.8a). The functions oflhcbm5,cab-8,lhcb5, andlhcbm8are related to the biosynthesis of some component proteins in PSII. The functions ofcpld35,fsd1, andprxqare related to the biosynthesis of enzymes involved in active oxygen metabolism.The functions ofggps,riba2,cartiso, andcrtrare related to the biosynthesis or regulation of some limiting enzymes in carotenoid or biosynthesis metabolism of biopigment. Among these genes, the extremely signif icantly diff erentially expressed geneggps is another prime molecular clone candidate gene because its relative signif icant expression change in the 3 comparison groups (P-value at least less than 0.01) measured by qPCR analysis (Table 4;Fig.8d).
3.8 Characterization of DsGGPS and DsDGAT
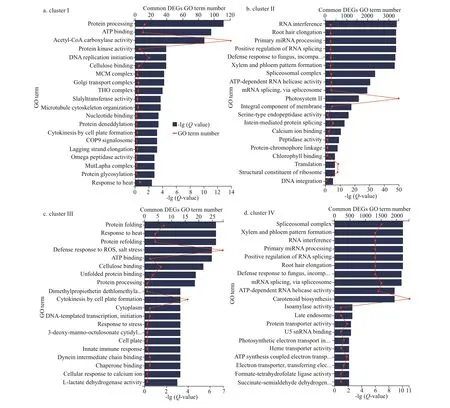
Fig.5 GO functional enrichment of the common DEGs divided into 4 clusters among the 3 comparison groups: DS_LS vs.DS_CO, DS_MS vs. DS_CO, and DS_HS vs. DS_CO
Based on the unigene information ofggps(unigene 47 314) from transcriptome sequencing, the key geneDsGGPS(encoding geranylgeranyl diphosphate synthase) inD.salina_YC was successfully cloned via RACE with specif ic PCR primers (Supplementary Table S7a & Fig.S6a). A full-length 1 876-bpDsGGPScDNA (MW=581.57 kDa) with a 220-bp 5′untranslated region (UTR) and a 582-bp 3′ UTR was assembled (Fig.9a). A 1 074-bp CDS was obtained for this gene with the same length as the predicted ORF.As a key enzyme in the biosynthesis of beta-carotene,the hydrophobic GGPS ofD.salina_YC (average value of hydrophobicity (Hphob./Kyte & Doolittle)was -0.297, as shown in Supplementary Fig.S6b)contained 357 amino acids, of which Ala accounted for the highest proportion (12.04%) and Cys accounted for the lowest proportion (1.96%). This predicted GGPS, with diverse amino acid sequences among plant species (Supplementary Fig.S6c), belongs to the isoprenoid biosynthesis C1 superfamily(Supplementary Fig.S6d) (Emmerstorfer-Augustin et al., 2016). Its pI value was 6.71 and MW value was 38.88 kDa inD.salina_YC. The putative 3D structure of GGPS inD.salina_YC was also predicted, with no signal peptides or transmembrane regions detected(Supplementary Fig.S6e). This GGPS appears to be a relatively conserved enzyme in the ML tree, with the highest homology among the fourDunaliellaspecies(Fig.9b).
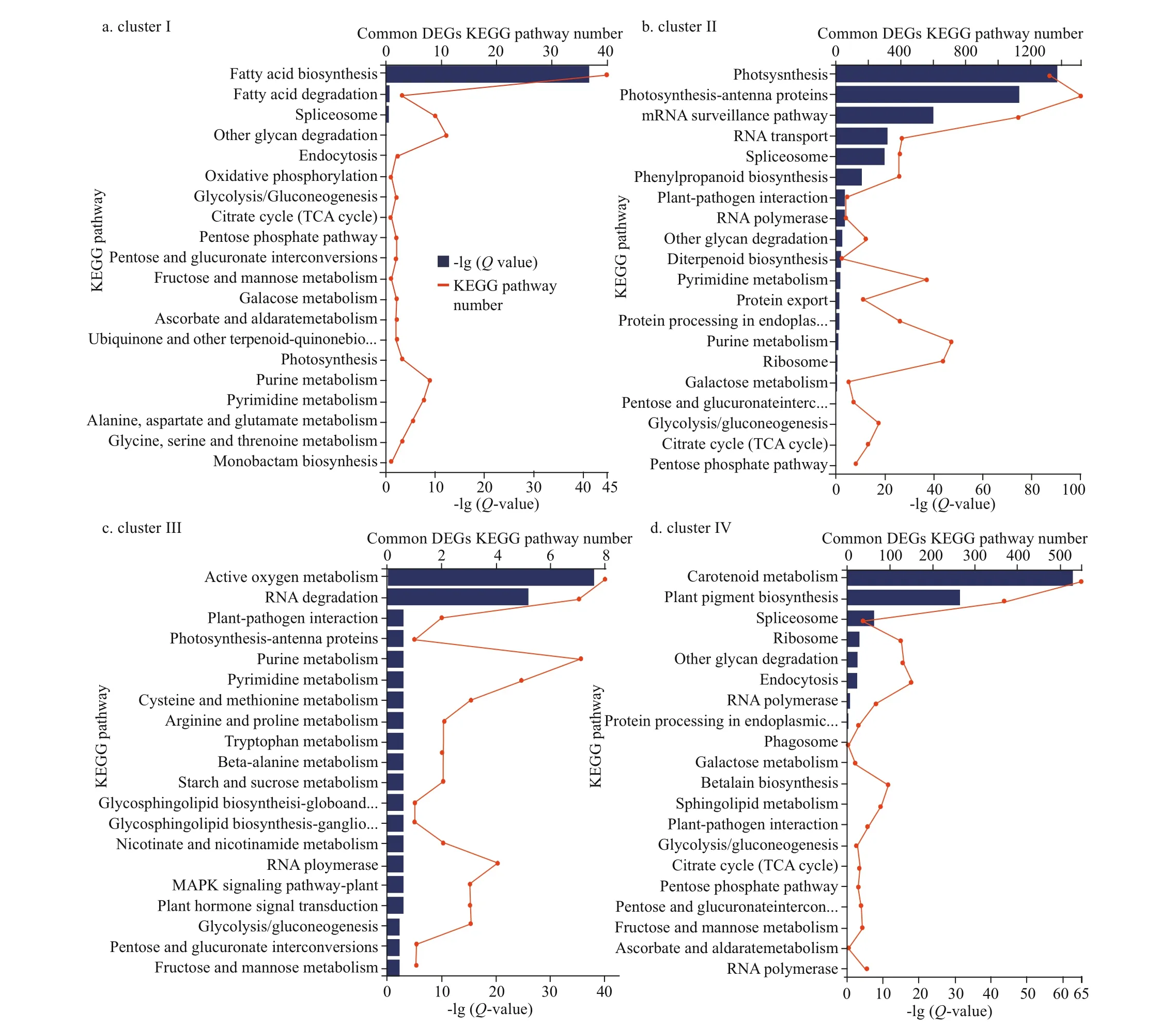
Fig.6 KEGG pathway functional enrichment of the common DEGs divided into 4 clusters in the 3 comparison groups:DS_LS vs. DS_CO, DS_MS vs. DS_CO, and DS_HS vs. DS_CO

Fig.7 Expression prof lies of the common DEGs in the 3 comparison groups: DS_LS vs. DS_CO, DS_MS vs. DS_CO, and DS_HS vs. DS_CO in the signif icantly enriched GO and KEGG pathways with 4 clusters, measured by the reads per kilobase of exon model per million mapped reads (FPKM) ratio of the experimental group to the the control
Based on the unigene information ofdgat(unigene 16 111), the key geneDsDGAT(encoding diacylglycerol O-acyltransferase) inD.salina_YC was also cloned via RACE with specif ic PCR primers(Supplementary Table S7b & Fig.S7a). A full length of 2 915-bpDsDGATcDNA (MW=918.64 kDa) with a 515-bp 5′ untranslated region (UTR) and a 756-bp 3′ UTR was assembled (Fig.9c). A 1 644-bp CDS was obtained in this alga with the ORFs predicted. The longest ORF was converted into a potential hydrophilic protein (the average value of Hphob./Kyte & Doolittle was 0.021, as shown in Supplementary Fig.S7b) with 547 amino acids(pI=9.51, MW=62.55 kDa). In the amino acids of this predicted protein, Leu had the highest (13.71%)proportion, and Cys had the lowest (1.83%)proportion; furthermore, the protein was predicted to encode DGAT inD.salina_YC, a key enzyme with diverse amino acid sequences among plant species(Supplementary Fig.S7c) that catalyzes the synthesis of triglycerides in the process of fatty acid biosynthesis and belongs to the membrane-bound O-acyl transferase (MOGAT) superfamily (Supplementary Fig.S7d) (Wang et al., 2013). The potential 3D model structure of this enzyme was also predicted, with no signal peptides or transmembrane regions detected(Supplementary Fig.S7e). The ML tree of DGATs showed that the amino acid sequence of DGAT inD.salina_YC was similar to those of DGATs in two green algae,C.zof ingiensisandRaphidocelissubcapitata(Fig.9d). The information obtained on both genes has been uploaded to GenBank in 2019 with the gene accession numbers MN172173 and MN172172.
3.9 Physiological and biochemical characterization
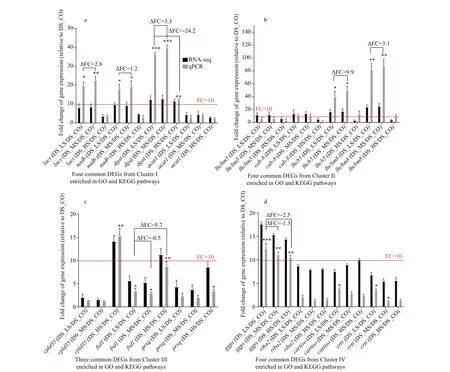
Fig.8 Comparative analyses of relative expression levels of the 15 common DEGs signif icantly enriched in GO and KEGG pathways
TLC, photochemical (PC) effi ciency, SSA, and BCC in the 3D.salina_YC experimental groups with DS_CO as the control were tested. These representative physiological/biochemical indexes could ref lect the main metabolic situations or biological processes of this alga that in the 15 validated genes involved. As Fig.10a and Supplementary Table S8a shown, the TLC ofD.salina_YC cells increased as salinity and growth time increased. However, the TLC of DS_HS was the highest (maximum TLC value=(1.53±0.30)×10-4mg/cell, all ratio values of TLC during the whole growth period relative to DS_CO>5). The maximum and potential PC effi ciencies ofD.salina_YC cells were inhibited with increasing salinity (Fig.10b & c;Supplementary Table S8b & c). The PSII primary PC effi ciency (Fv/Fm) of DS_HS, DS_MS, and DS_LS was less than half within 3, 9, and 31 d, respectively(Fig.10b). The PSII potential PC activity (Fv/Fo) of DS_HS was less than half within 46 d (Fig.10c). By contrast, the PC effi ciency of DS_CO was the least aff ected (ΔFv/Fm=0.11, ΔFv/Fo=0.94). Unlike the ratio of SSA, which exhibited slight f luctuations among the 3 algal comparison groups (relative to DS_CO), the SSA of theD.salina_YC cells at their mature phase was enhanced with an increasing NaCl concentration and prolonged growth time (Fig.10d, Supplementary Table S8d). The maximum SSA value reached 0.22±0.01 U/mg in DS_HS at the mature phase at 60 min, with approximately 11 times higher SOD specif ic activity relative to that in DS_CO. Moreover,based on the beta-carotene extracts concentration(Supplementary Fig.S8, Tables S8e), both the BCC ofD.salina_YC cells under high-salinity stress(maximum BCC value was 13.34±0.35 mg/cell on day 51) and their ratio relative to the control were the highest (maximum ratio of BCC relative to control was 8.8±0.49 on day 46) among the 3D.salina_YC comparison groups (Fig.10e).

Fig.9 Sequences analyses of the geranylgeranyl diphosphate synthase and diacylglycerol O-acyltransferase coding gene in D. salina_YC ( DsGGPS and DsDGAT), and prediction of their encoded proteins
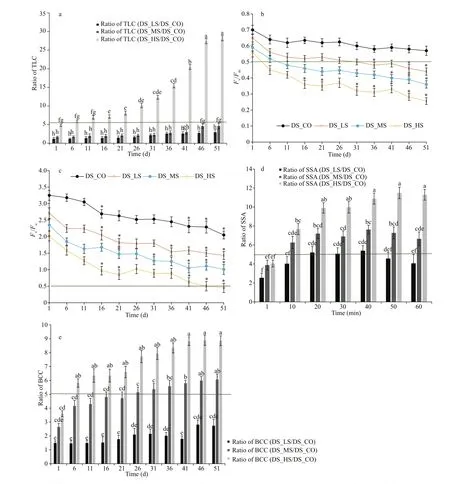
Fig.10 Comparative analyses of physiological and biochemical indexes in D. salina_YC under diff erent salinity stresses
4 DISCUSSION
Shetty et al. (2019) reported that the low salinity condition (0.05-0.15-mol/L NaCl) increased the photosynthetic activity ofD.salinaover a short period of time but that higher salinity (0.3-0.4-mol/L NaCl) decreased it over a long period of time. The medium salinity, 0.5-mol/L NaCl, used as the stress condition for transcriptomic analysis ofD.salinaduring its stationary phase revealed 4 core elements of theDunaliellasalt stress response system (Panahi et al., 2019). Another transcriptomic analysis of
D.salinawith a high-salinity treatment (2.5-mol/L NaCl) for 2 h revealed a large number of upregulated genes involved in 7 diff erent metabolic or biological processes (He et al., 2020). Based on the above f indings, we found that a more detailed salinity gradient treatment would be essential for revealing much more interesting functional genes and physiological features in this alga. Herein, 0.51-,1.03-, and 3.42-mol/L NaCl concentrations were used as the abiotic stress conditions with 0.05-mol/L NaCl concentration as a control condition for transcriptomic analysis of this unique high-salt-tolerantDunaliellaspecies from Yuncheng Salt Lake.
Recently, transcriptomic analysis has helped elucidate the complexity of gene regulation under salt stress condition in algae. In this study, theD.salina_YC RNA-seq data per algal sample (mean value of raw reads: 73.9 M, mean value of clean reads:68.6 M, GC mean content: 51.84%) obtained from the BGISEQ-500 platform were more abundant than the RNA-seq data ofD.salina(value of raw reads:38.5 M, clean reads: 31.2 M, GC content: 50%)obtained with the Illumina GAIIx platform (Panahi et al., 2019). Moreover, each cDNA library ofD.salina_YC exhibited a Q20% ranging from 97.42%to 97.67%, which are close to the percentages reported forD.salinastrain CCAP19/18 andD.salinastrain CCAP 19/30 (Polle et al., 2017; He et al., 2020). The number of CDSs inD.salina_YC based on BGISEQ-500 was 4 9751, ranging from 297 to 11 943 bp in length, which is higher than that generated forD.salinaCCAP 19/30 with the Illumina HiSeq 2000 system (43 864 CDSs, ranging from 242 to 8 978 bp) (He et al., 2020). Furthermore, an average of 9 681 unigenes were obtained from eachD.salina_YC sample in this study, which is greater than the individual gene number ofD.salinaCCAP 19/30 reported above (9 256 genes). However, the average length (856 bp) of contigs inD.salina_YC with a 1 323-bp N50 was less than that inD.salinastrain 435 (an average contig length of 1 409 bp, N50:1 727 bp) (Hong et al., 2017). In summary,BGISEQ-500 is an eff ective next-generation highthroughput sequencing platform. The transcriptome sequencing results ofD.salina_YC generated with this platform are high-conf idence.
The TLC ofD.salinacells may be related to the activities of enzymes that catalyze fatty acid biosynthesis or degradation (Af ify et al., 2010).However, this correlation changes due to the corresponding genes that are diff erentially expressed in this alga under diff erent salinity conditions. The present study showed that the TLC ofD.salina_YC progressively increased with global high DEGs expression levels (FPKM mean ratio of 8.27 relative to the control) when the salinity was 0.51-1.03 mol/L(Fig.7a). In this salinity range, 3 validated genes (lacs,dgat, andacat1) encoding the enzymes that catalyze fatty acid biosynthesis (Figs.5a & 6a; Table 4) were upregulated with positive changes in FPKM and qPCR values (Fig.11a & b). However, another validated gene,nadb, regulated fatty acid degradation and was downregulated with negative changes measured by FPKM. When the salinity reached 3.42 mol/L, the TLC ofD.salina_YC was markedly increased (a maximum value of 5.4231E-05 mg/cell)(Fig.10a), with most of the DEGs downregulated(FPKM mean ratio of 2.23 relative to the control)(Fig.7a). At the moment, all 4 validated genes were repressed based on their negative changes via FPKM and qPCR changes analyses (Fig.11a & b). We speculate that other lipid metabolic enzymes may induce fatty acid biosynthesis and inhibit its degradation processes. Hypersalinity stress may mediate enhancement of glycerol biosynthesis in this alga, which would accelerate fatty acid accumulation in algal cells to synthesize a large amount of lipids.Rismani-Yazdi et al. (2011) proposed a similar hypothesis in another halophilic alga,D.tertiolecta,using transcriptomic analysis.
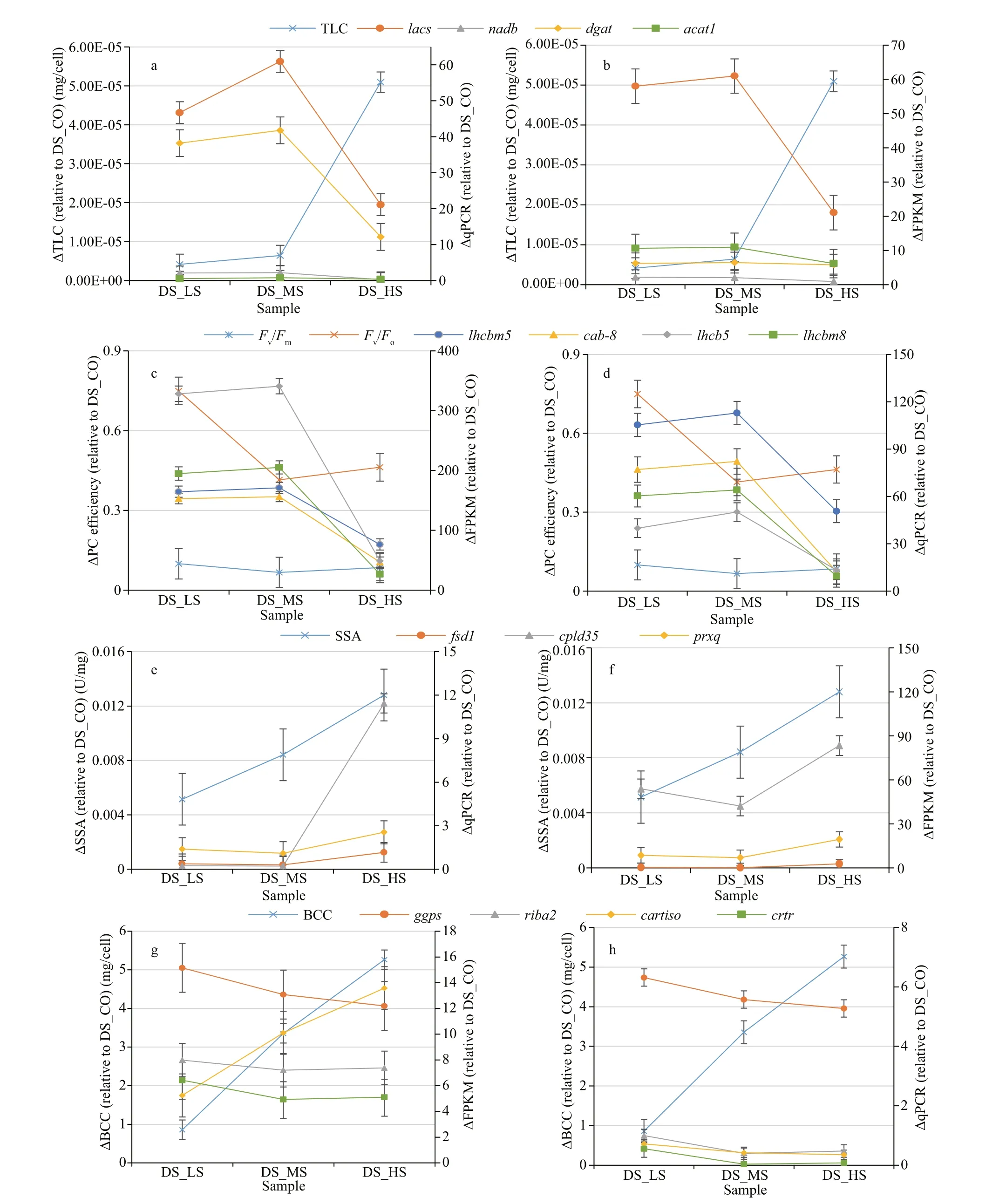
Fig.11 Relative change levels of total lipid content (TLC), PC, SOD specif ic activity (SSA), and beta-carotene content (BCC),and their corresponding validated DEGs in 3 D. salina_YC comparison groups relative to DS_CO measured by FPKM (b, c, f, g) and qPCR (a, d, e, h)
Much fewer algal cells with scarce contents in the small intracellular space were observed in theD.salina_YC control group with 0.05-mol/L NaCl treatment (Table 1; Fig.1a). The PC effi ciency (Fv/Fmranged from 0.70 to 0.57, andFv/Foranged from 3.25 to 2.05) decreased slowly in the control group (very low salinity condition) compared with the higher salinity stress groups (Fig.10b & c). Some studies showed that, with the salt-stress going on,photosynthesis or chlorophyll biosynthetic rates decreased inD.salina(Shetty et al., 2019; He et al.,2020), which indicates the consistent trend with PC in this alga. Relative to the control, the negative change in PC effi ciency in the 3 comparison groups increased with increasing algal salinity (not greater than 3.42 mol/L) (Fig.11c & d). Under low and medium salinity conditions, the global expression level of photosynthesis-related DEGs inD.salina_YC was high (FPKM mean ratio of 16.1 relative to the control)(Fig.7b). The 4 validated genes (lhcbm5,cab-8,lhcb5,andlhcbm8) encoded light-harvesting proteins or chlorophylla/b-binding proteins (Figs.5b & 6b;Table 4), which are involved in algal PC reactions and are upregulated with positive changes measured by FPKM and qPCR as the salinity increased by no more than 3.41 mol/L (Fig.11c & d). However, when the salinity reached 3.41 mol/L, the negative change in PC effi ciency increased with the related DEGs overall downregulated (FPKM mean ratio of 6.89 relative to the control) (Fig.7b). Meanwhile, the 4 validated genes above were markedly repressed (Fig.11c & d)based on FPKM and qPCR changes analyses, which indicates that hypersalinity stress may inhibit gene expression involved in the absorption and transition of light inD.salina_YC photosynthesis. Zhou (2013)proposed a similar hypothesis inZosteramarinaL.using qPCR analysis.
The SSA ofDunaliellashould be a very important parameter for investigating the algal response to salt stress-induced ROS damage (Qv and Jiang, 2013).The SSA ofD.salina_YC increased (maximum value of 0.022 U/mg) in 60 min with increased environmental salinity in this study (Supplementary Table S8d). The global expression level of ROS response-related DEGs in this alga was high (FPKM mean ratio of 8.87) under 3 diff erent salinity stresses relative to the control (Fig.7c). However, the three validated genescpld35,fsd1, andprxqencoding oxidoreductase-like protein, Fe-SOD and peroxidase (or its precursor),respectively in this alga (Figs.5c & 6c; Table 4), were downregulated with a salinity increase of no more than 1.03 mol/L and then upregulated with increasing salinity via RNA-seq and qPCR changes analyses(Fig.11e & f). The 4 genes underwentD.salina_YC positive ROS response regulation when it suff ered from hypersalinity stress (Fig.11e & f). Panahi et al.(2019) obtained similar results inD.tertiolectabased on the ROS scavenging-related genesapxandshmt2.Moreover, the genecpld35was markedly upregulated inD.salina_YC with a salinity increase of more than 1.03 mol/L measured by FPKM and qPCR changes.We speculate that this gene, encoded an oxidoreductaselike protein that could play an essential role in the regulatory progress of ROS scavenging in this alga especially under extreme salinity stress condition.
Alkayal et al. (2010) determined that hypersalinity(greater than 2.5-mol/L NaCl) triggered the synthesis of many bioactive substances or metabolites inD.salina. The BCC ofD.salina_YC increased with increasing salinity (average increment of 5.27 mg/cell relative to the control), especially 3.42-mol/L NaCl(Supplementary Table S8e), with enrichment of intracellular yellow-green pigments (Fig.1). A similar situation was found in theD.salinaTeod (Ramos et al., 2011). The common DEGs in the 3D.salina_YC salt-stress comparison groups involved in the carotenoid biosynthesis were downregulated overall(FPKM mean ratio of 5.18 relative to the control)(Fig.7d), with two validated genes,riba2andcrtrencoding the ribof lavin biosynthesis protein (possible beta-carotene synthesis-related non-key factor) and beta-carotene hydroxylase (a key enzyme that catalyzes the conversion of beta-carotene to zeaxanthin), respectively (Figs.5d & 6d; Table 4),were slightly downregulated when the salinity was 0.51-1.03 mol/L, and then slightly upregulated when the salinity continued to increase measured by FPKM and qPCR changes (Fig.11g & h). This gene regulation pattern involved in carotenoid biosynthesis was also shown in another green alga,Chlorellasp., following salt stress treatment (Li et al., 2018). Moreover, the geneggpsencoding geranylgeranyl diphosphate synthase was continued downregulated with salinity increasing. Conversely, the upregulated expressed genecartisomeasured by FPKM encoding carotenoid isomerase with salinity increasing may play a positive regulatory role for BCC accumulation inD.salina_YC. A similar conclusion was reached for another green alga,C.zof ingiensis(Mao et al., 2020).Overall, low or medium salinity conditions may be a positive factor for rapid cellular proliferation, active substance accumulation and related genes expression inD.salina(Talebi et al., 2013). Very low salinity should be a basic factor for maintaining live algae.Although high salinity may be a negative factor for the growth of this alga, a few unique bioactive substances or high-value-added metabolites may be obtained from it. Finally, we would like to acknowledge the limitations of this study. Although some experimental methods have been utilized to explore the potential salt tolerance or response mechanisms inD.salina_YC, regulation at the posttranscriptional, translational and protein levels cannot be ignored. Gene knockout and overexpression technologies could be used for expression and functional validation of some key saltstress related genes inD.salina. From this point of view, it is necessary to further illustrate the detailed molecular mechanism underlying the response to salt stress inD.salinabased on these candidate key genes,which will be helpful to our protection ofDunaliellacommunity and the effi cient utilization of unique species in the future.
5 CONCLUSION
This study provided an overview of the diff erent responses ofD.salina_YC to 4 diff erent NaCl concentrations at the transcriptomic level combined with the morphological, physiological, and biochemical levels. Fifteen common DEGs among the total unigenes of the fourD.salina_YC experimental groups used for sequencing were signif icantly enriched and are possibly involved in enhancing lipid metabolism, decreasing photosynthesis, constantly increasing active oxygen metabolism and accumulating carotenoids. Diff erential expression and cloning of some genes at the molecular level will help uncover the mechanism underlying the response or tolerance ofD.salina_YC to diff erent salinity environments in Yuncheng Salt Lake.
6 DATA AVAILABILITY STATEMENT
The raw datasets of transcriptome in the study are available from the NCBI Short Read Archive (SRA)database with the accession numbers SRR12326540,SRR12326982, SRR12327191, SRR8393723,SRR8393722, SRR8393725, SRR8393724,SRR8393727, SRR8393726, SRR8393729,SRR8393728, and SRR8393721. The authors declare that all the other data supporting the f indings of this study are available within the article and its supplementary f iles.
7 AUTHOR CONTRIBUTION
Fan GAO performed the experiments, analyzed the data, and co-wrote the manuscript; Fangru NAN, Jia FENG, Junping LÜ, and Xudong LIU analyzed the data; Shulian XIE supervised the project and revised the manuscript. All authors have read and approved the f inal manuscript.
8 ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All sampling procedures in this experiment were in accordance with the guidelines for the care and use of laboratory plants of Shanxi University and were approved by the Wildlife Care and Use Committee of Yuncheng Forestry Bureau, Shanxi Province, China.
杂志排行

Journal of Oceanology and Limnology的其它文章
- MamZ protein plays an essential role in magnetosome maturation process of Magnetospirillum gryphiswaldense MSR-1*
- Maximum sustainable yield estimation of enhancement species with the characteristics of movement inside and outside marine ranching*
- Neglectella glomerata sp. nov., a new species and implications for the systematics of the genus Neglectella (Oocystaceae,Trebouxiophyceae, Chlorophyta)*
- A new species Lobophora tsengii sp. nov. (Dictyotales;Phaeophyceae) from Bach Long Vy (Bailongwei) Island,Vietnam*
- Characterization and diversity of magnetotactic bacteria from sediments of Caroline Seamount in the Western Pacif ic Ocean*
- Evidence for the role of KIF17 in f ish spermatid reshaping:expression pattern of KIF17 in Larimichthys polyactis spermiogenesis*