MR‐Egger 回归在孟德尔随机化分析中的应用
2021-12-07徐艺耘刘振球施婷婷张铁军
徐艺耘 刘振球 樊 虹 张 欣 施婷婷 吴 声 张铁军,2△
(1复旦大学公共卫生学院流行病学教研室 上海 200032;2复旦大学义乌研究院 义乌 322000)
孟德尔随机化(Mendelian randomization,MR)是借助遗传变异(genetic variation)作为工具变量(instrumental variable,IV)来推断暴露因素与结局之间因果关联的方法,有效避免了反向因果关联和潜在混杂因素导致的偏倚。随着全基因组关联研究(genome-wide association study,GWAS)的增多,孟德尔随机化分析广泛应用于观察性流行病学研究中。传统孟德尔随机化分析方法,如逆方差加权法(inverse-variance weighted,IVW),假定所有的遗传变异均满足工具变量的3 个核心假设[1]:(1)工具变量与暴露因素之间有强相关(关联性假设);(2)工具变量与混杂因素无关联(独立性假设);(3)工具变量只能通过暴露因素对结局产生作用,不能通过其他途径影响结局发生(排他性假设)。当工具变量存在多效性时,因果效应的估计则会有偏[2]。MR-Egger 回归是在IVW 的基础上修正而来的一种基于汇总数据的多工具变量孟德尔随机化方法。与IVW 不同的是,该方法仅需满足工具变量多效性效应独立于工具变量与暴露因素之间的关联(instrument strength independent of direct effect,InSIDE)假设和无测量误差(no measurement error,NOME)假设,不如工具变量的3 个核心假设要求严格。同时,该方法既能检测多效性又能校正多效性偏倚,所以在以多个遗传变异作为工具变量的研究中,采用MR-Egger 回归能够保持孟德尔随机化方法的有效性。 本研究将对MR-Egger 回归进行阐述,并用此方法探索高密度脂蛋白胆固醇(highdensity lipoprotein cholesterol,HDL-C)与冠状动脉疾病(coronary artery disease,CAD)之间的因果关联。
资料和方法
基本原理MR-Egger 回归是由Bowden 等[3]于2015年提出,广泛运用于采用多个单核苷酸多态性(single nucleotide polymorphism,SNP)作为工具变量进行因果推断的孟德尔随机化分析中,尤其适用于遗传变异存在定向多效性的情况。该方法的核心在于加权线性回归时考虑截距项的存在,利用截距项来衡量工具变量间平均多效性的大小,斜率则是因果效应的无偏估计。MR-Egger 回归的效用取决于两个前提:第一,InSIDE 假设要求SNP 对暴露因素的影响独立于它们对结局的多效性影响,弱化了排他性假设;第二,NOME假设要求工具变量与暴露因素的关联中不存在测量误差,I2统计量可以评估是否满足NOME 假设,如果不满足,则会产生弱工具变量偏倚[4]。
记J 个工具变量为Gj(j=1,2,…,J),暴露因素为X,结局为Y,工具变量Gj 对暴露因素X 和结局Y的效应估计值分别为β̂Xj和β̂Yj及其对应的标准误为和,遗传变异与结局之间的关联可表示为:

其中,αj 表示工具变量Gj 对结局Y 的直接影响,当αj≠0 时,工具变量具有多效性;θ 表示暴露因素X 对结局Y 的影响;θβXj则为工具变量Gj 通过暴露因素X 对结局Y 的间接影响[5]。
对于满足工具变量所有假设的单一遗传变异Gj(αj = 0),暴露因素X 对结局Y 的因果效应可被一致性地估计为关联估计的比值,比值及相应的标准误分别为:。
由于MR-Egger 回归并没有限制截距项为0,回归方程可表示为:


当InSIDE 假设和NOME 假设均满足时,θ是因果效应的无偏估计[6]:

若满足InSIDE 假设但不满足NOME 假设,因果效应估计值则会有偏:
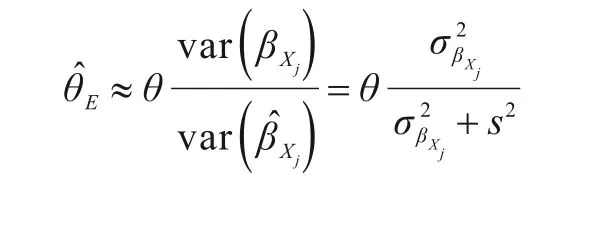
其中,s2为测量误差,s2= 0 时才满足NOME 假设;I2统计量是在Meta 分析中评估研究间异质性的工具,的大小可用来估计,代表SNP-暴露关联的真实方差,取值范围为0~1,越接近1,MR-Egger回归的估计越接近真实因果效应。其中,Cochran’s Q 统计量是评估工具变量间异质性的工具,是SNP-暴露关联的平均值。
IVW 法是由Burgess 等[2]于2013年提出的一种加权线性回归模型。与MR-Egger 回归不同,该方法限制回归直线通过原点,将两个或多个SNP 的效应值进行整合,因果效应估计值为:
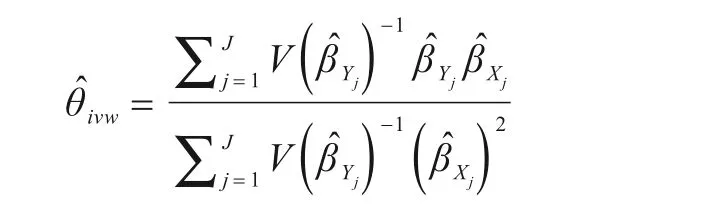
实例分析HDL-C 资料来自全球脂质遗传学(Global Lipids Genetics Consortium,GLGC)数据库(http://csg. sph. umich. edu/willer/public/lipids2013/),该数据库中有关基因位点与HDL-C的GWAS 研究结果发表于2013年,该研究样本量为94595 人,分析了2418527 个与HDL-C 相关的SNP 位 点[7]。 与HDL-C相关的SNP筛选标准:(1)达到全基因组统计显著性水平,即P<5×10-8;(2)去除连锁不平衡(linkage disequilibrium,LD)的SNPs,排除标准为r2>0.01,kb= 5000,筛选出125个与HDL-C 相关且相互独立的SNP,提取每个SNP 的rs 编号、效应等位基因(effect allele,EA)和次要等位基因(non-effect allele,non-EA)、β 系数、P值和标准误。 CAD 数据来源于2018年Van Der Harst 等[8]发表的GWAS 统计结果,该研究将CARDIoGRAMplusC4D[Coronary Artery Disease Genome wide Replication and Meta-analysis(CARDIoGRAM) plus The Coronary Artery Disease(C4D)Genetics]的数据与英国生物银行(UK Biobank,UKBB)的CAD 数据进行GWAS分析,共包括547216 人,其中病例组122733 人,对照组424528 人,分析了69033 个与CAD 相关的SNP位点。 设置最小等位基因频率MAF=0.01,从GLGC 数据库筛选的SNP 在CAD 来源的GWAS研究中均存在,经等位基因对齐后,rs11637365、rs3790106、rs4986970、rs6589581 和rs964184 由于存在回文结构被剔除,最终有120 个SNP 作为工具变量分析HDL-C 与CAD 的因果关联。上述研究人群均为欧洲血统,具有相似的年龄和性别,符合两样本孟德尔随机化分析的要求。 利用R4.0.3 中的TwoSampleMR 包进行两样本孟德尔随机化分析,检验水准α=0.05。 因果效应分析使用IVW 法和MR-Egger 回归,对比两者差异;敏感性分析包括利用MR-Egger 回归进行多效性检验(pleiotropy test)和逐个剔除检验(leave-one-out sensitivity test),“leave-one-out”是指逐一剔除SNP,分别计算剩下SNP 的合并效应,如果剔除某个SNP 后其他SNP估计出来的MR 结果和总结果相差很大,说明MR结果对该SNP 是敏感的。
结果
本研究针对HDL-C 和CAD 选取有效SNP 作为IV,经筛选确定了120 个SNP 作为IV,表1 列出了前10 个SNP 的相关信息。采用MR-Egger 回归和IVW 法估计因果效应(表2)。 IVW 结果支持HDL-C 和CAD之间存在因果关系(OR=0.82,95%CI:0.75~0.89),且HDL-C 每增加1 个标准差,CAD 风险降低18%。而MR-Egger 法不支持(OR=0.96,95%CI:0.83~1.11),即HDL-C 对CAD 的发生风险没有影响。多效性检验结果显示,MR-Egger回归的截距为-0.01,P=0.008,说明因果分析结果会受到水平多效性的影响。在水平多效性存在的情况下,传统孟德尔随机化方法IVW 的估计出现了偏差,MR-Egger 回归则能识别并校正多效性,给出接近真实值的因果估计。敏感性分析结果见图1,所有线条均在0 的左侧,说明无论去除哪个SNP 都不会对结果产生根本影响,即孟德尔随机化的结果稳健。

表1 工具变量SNP 信息Tab 1 Information of the instrumental variable SNP

表2 因果效应估计Tab 2 Estimation of causal effects

图1 逐个剔除检验Fig 1 Leave‐one‐out sensitivity analysis
讨论
早期孟德尔随机化研究倾向于使用单一遗传变异,并集中在单一研究人群中的特定危险因素与疾病的关联研究。近年GWAS 中大量增长的基因型-表型关联导致众多暴露因素的大量遗传变异被识别出来[9]。许多遗传变异被认为具有多效性效应,要证明排他性假设的成立几乎不可能。 MREgger 回归区别于传统孟德尔随机化方法之处在于放宽了SNP 之间不存在水平多效性的要求,它假设在基因-暴露关联和基因变异对结果的直接影响之间没有相关性,是比更严格的排他性标准更弱的要求,即使所有工具变量是无效的,MR-Egger 回归也能得到准确的因果效应估计[10]。因此,在工具变量存在多效性的情况下,MR-Egger 回归是一个更好的选择。
本研究借助GLGC 数据库和Van Der Harst 的GWAS 统计结果,提取与HDL-C 和CAD 均密切相关且相互独立的SNP 作为工具变量,利用MREgger 回归和IVW 法进行两样本孟德尔随机化分析,估计HDL-C 和CAD 的因果关联并进行比较。IVW 结果显示两者之间具有因果关系,HDL-C 对于CAD 有保护作用,但IVW 法假设截距为0,未考虑所纳入的工具变量的基因多效性;而MR-Egger回归并不局限于截距为0,是在IVW 基础上的修正,同时考虑基因多效性对结果的影响,因此给出了并不一致的估计结果,即HDL-C 与CAD 之间不存在因果关联,与以往孟德尔随机化研究结果相吻合[11]。随后的逐一剔除检验显示MR-Egger 结果更具有稳健性。因此,MR-Egger 回归在孟德尔随机化分析中有较好的应用价值。
与传统孟德尔随机化方法相比,MR-Egger 回归的偏倚更小,而且因果无效假设的排斥率更接近名义上的5%[3]。MR-Egger 回归的局限性是它在因果估计方面的统计效力较低。如果没有证据表明IVW 和MR-Egger 之间存在系统性差异,那么通常采用IVW,虽然IVW 可能不那么稳健,但在此情况下,IVW 估计值的精度更高。有研究者指出MREgger 回归应被视为一种敏感性分析,用来检验是否违反工具变量假设,而不是作为上述更常见方法的替代[12]。实际应用时应结合多种方法的结果进行综合考量,如果多种方法的结果一致,则结论更为可靠,从而更好地应用于传统观察性流行病学研究,为病因推断提供更强有力的证据,并有利于为未来疾病预防的干预目标设定优先级。
作者贡献声明徐艺耘 查阅文献,论文构思、撰写和修订,数据统计分析。刘振球,樊虹,张欣,施婷婷,吴声 结果解释,论文修改。 张铁军论文构思、指导和修订。
利益冲突声明所有作者均声明不存在利益冲突。
