三元硫族半导体化合物K2Zn3S4 的制备与光学性质研究
2021-12-02王能能姜昱丞姚金雷
彭 涛, 王能能, 姜昱丞, 赵 蒙, 姚金雷*
(1.苏州科技大学 物理科学与技术学院,江苏 苏州 215009;2.苏州科技大学 江苏省微纳热流技术与能源应用重点实验室,江苏苏州 215009)
透明导电材料不仅具有良好的导电特性而且对可见光有高的透过率,广泛应用于太阳能电池、显示器、发光二极管等器件中。 工业应用的透明导电材料大部分是氧化物,比如Sn 掺杂In2O3[1]、F 掺杂SnO2[2]和Al 掺杂ZnO[3]。但是这些透明导电氧化物一般是n 型半导体,p 型掺杂很难实现。近年来的报道表明硫族半导体也可作为性能优良的透明导电材料来研究,而且它们大部分都呈现p 型导电特性。 表现出独特透明导电特性的硫族半导体包括二元硫化物的ZnS、CdS、ZnxCd1-xS、SnS2、In2S3等[4-6],三元化合物的CuAlS2[7-9]、α-BaCu2S2[10]、Cu3TaS4[11]等,以及混合阴离子化合物的LaCuOS、BaCuSF、CuSCN 等[12-13]。
新型的透明导电材料必须具备大的光学带隙(Eg>3.1 eV)、高载流子迁移率、低载流子浓度、在宽光谱范围内有高透光率等特性。为了发展新型的硫族透明导电半导体,Androulakis 等人[14]提出利用大尺寸原子插入已知硫族化合物的结构单元之间,增大其光学带隙从而得到透明导电材料的想法。 在零带隙的二元半导体化合物HgSe 中,Androulakis 等人把大尺寸的碱金属Cs+离子嵌入[HgSe4]之间,随着插入Cs+的增多,晶体结构发生维度的降低且光学带隙也在逐渐增大:三维网状结构的HgSe(Eg=0 eV)→开放式三维结构的Cs2Hg6Se7(Eg=1.0 eV)→二维层状结构的Cs2Hg3Se4(Eg=2.1 eV)→一维链的Cs2HgSe2(Eg=3.0 eV)→零维分子单元的Cs6HgSe4(Eg>4 eV),从而在硫族半导体中合成出新的透明导电材料。 由于碱金属或者碱土金属离子的电负性、原子尺寸等性质与过渡金属元素有显著不同,元素的替代会引起配位环境和能带结构的变化,从而得到丰富的晶体结构和物理性能的变化。 在ZnSe→Cs2Zn3Se4[15]、ZnTe→Cs2Zn3Te4[15]、ZnS→Cs2Zn3S4[16]、CdTe→(Rb,Cs)2Cd3Te4[17]等取代中都发现了新型硫族材料的存在,表现出独特的半导体特性。 其中,Cs2Zn3Se4和Cs2Zn3Te4化合物的带隙分别为3.61 eV 和2.83 eV,展现出优良的透明导电性能。
文中,笔者选取典型的二元硫化物ZnS 作为研究对象,利用碱金属K 插入ZnS 结构中,以期得到新型的三元硫族半导体化合物,并系统研究该化合物的晶体结构和光学性能之间的关系。
1 实验和计算
1.1 单晶生长
文中用于单晶生长的原料包括K2S 粉末(RG,Acros)、Zn 金属颗粒(99.995%,中诺新材科技有限公司)和S 粉末(99.999%,中诺新材科技有限公司)。 为了避免材料氧化和吸潮,所有原料的称量、研磨和混合都在充氩气的手套箱内进行。 K2Zn3S4的单晶通过助溶剂法使原料溶解后自发结晶生长,具体实验过程如下:
(1)在手套箱内称量K2S(3 mm,0.331 0 g)、Zn(6 mm,0.392 4 g)和S(6 mm,0.192 0 g),将称量好的原料在玛瑙研钵中研磨2 min,随后倒入石墨坩埚(Φ14 mm,高20 mm,壁厚3 mm),盖上石墨盖后转移至25 mL的高温高压釜内,手动旋紧后移出手套箱,再用大的活动扳手旋紧。
(2)高压釜转移至马弗炉内进行晶体生长,从室温以1 ℃·min-1升温至850 ℃,并在850 ℃保温两天,再以4 ℃·h-1降至650 ℃后,以15 ℃·h-1降温至50 ℃,最后断电自然冷却至室温。
反应结束后,将高压釜旋开取出石墨管,在通风橱里将其砸开,捡出反应物放入烧杯中,用N,N-二甲基甲酰胺(DMF,99.5%,Aladdin)在磁力搅拌器上清洗助溶剂,接着用酒精冲洗3 遍后晾干,得到黄色透明的K2Zn3S4晶体。 样品在空气中是稳定的。
1.2 结构和性能表征
把少量单晶样品碾碎成粉末,利用Bruker D8 Advance 型X 射线衍射仪(Cu-Kα 射线,λ=0.154 184 nm)收集粉末样品的X 射线衍射谱(XRD)。利用扫描电子显微镜(SEM,Hitachi SU8010 型)和能量色散X 射线光谱(EDAX,Octane Plus 型)表征单晶样品的表面形貌和化学组成。采用LabRAM HR Evolution 显微共焦拉曼光谱仪测量样品的拉曼谱,使用532 nm 的绿光作为激发光源,功率约为60 mW,收集范围为50~100 0 cm-1。 红外光谱使用Bruker VERTEX 70 红外光谱仪测量,波数测量范围为600~400 0 cm-1,分辨率2 cm-1。 红外样品是将待测样品与干燥的KBr 按照1∶100 的比例均匀混合,然后压成透明的薄片而得到的。样品的漫反射光谱由Shimadzu UV-2600 型紫外-可见-近红外分光光度计测得, 该仪器的双光路配置了积分球, 测量范围为λ=200~200 0 nm。
1.3 第一性原理计算
为了研究样品的晶体结构和光学性质之间的关系,笔者利用第一性原理计算了它们的能带结构和拉曼谱,采用Materials Studio 软件的CASTEP 模块计算[18],具体为:
(1)能带结构计算:首先利用XRD 数据解析出的晶体结构作为输入模型;然后利用BFGS 方法进行几何构型优化,晶胞参数和各原子占位均放开弛豫;在结构优化的基础上,采用超软赝势和广义梯度近似方法(GGA+PBE)的交换关联泛函进行电子能带结构计算,平面波截断能量为480 eV,自洽场SCF 收敛的计算精度设为5×10-7eV。
(2)拉曼光谱计算:以上步结构优化得到的晶体结构为模型,利用模守恒(Norm conserving)赝势和有限位移法(Finite displacement)计算拉曼谱,平面波截断能量为720 eV,SCF 收敛的计算精度设为5×10-7eV。
2 结果与讨论
2.1 结构分析
图1 为K2Zn3S4的粉末XRD 谱, 与文献报道的K2Mn3S4型结构(单斜晶系的P2/c,No.13)的模拟XRD谱吻合,说明文中样品的晶体结构与K2Mn3S4同型。 利用Rietica 软件对XRD 数据进行了Rietveld 精修,得到K2Zn3S4的 晶 胞 参 数 分 别 为:a=0.716 47(5) nm、b=0.573 77(3) nm、c=1.087 82(6) nm、β=112.186(4)°和V=0.414 08(4) nm3,与文献报道的数据较为接近[19]。相应的原子占位列于表1,共价键长列于表2 中。
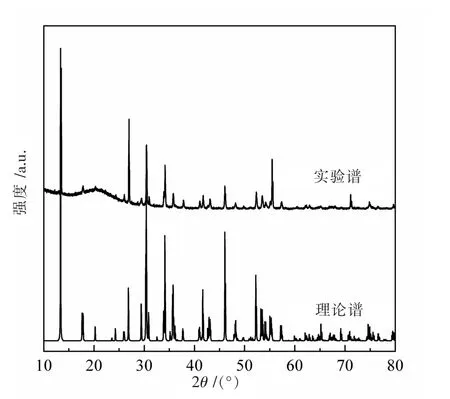
图1 K2Zn3S4 的粉末XRD 谱

表1 K2Zn3S4 的原子坐标(空间群P2/c,Z=2)

表2 K2Zn3S4 晶胞的原子间距
K2Zn3S4的晶体结构如图2 所示,从图2(a)可看到K2Zn3S4具有二维层状结构,K 离子沿着x 轴填充在[ZnS4]层之间。 在y-z 平面,[ZnS4]四面体通过共享边相互连接,构成[ZnS4]二维层,四面体未铺满整层,有空隙存在(图2(b))。原子Zn 周围有4 个近邻的S 原子,构成了[ZnS4]四面体的配位环境,这种配位结构在多种硫化物中都能观察到[20]。 原子Zn1 有两个不等长的Zn-S 键,为0.230 55 nm 和0.252 16 nm;原子Zn2 的Zn-S键长处于0.223 42~0.259 33 nm,都互不等长;这说明Zn 具有畸变的四面体配位环境(见表2)。 每个K 原子都有8 个最近邻的S 原子,但K-S 键并不等长,介于0.330 69~0.350 88 nm之间,构成了伪立方体的[KS8]配位环境。 在多元硫化物中,K原子常有[KS6]八面体、[KS7]单帽三角棱柱、[KS8]双帽三角棱柱等多配位体环境[21-22],这与K 具有较大的原子尺寸相关。 这些伪立方体[KS8]通过共享边相互连接,铺满了整个y-z 平面;它们和[ZnS4]二维层沿着x 轴通过共享边相互连接,构成了整个晶体结构(图2(c))。
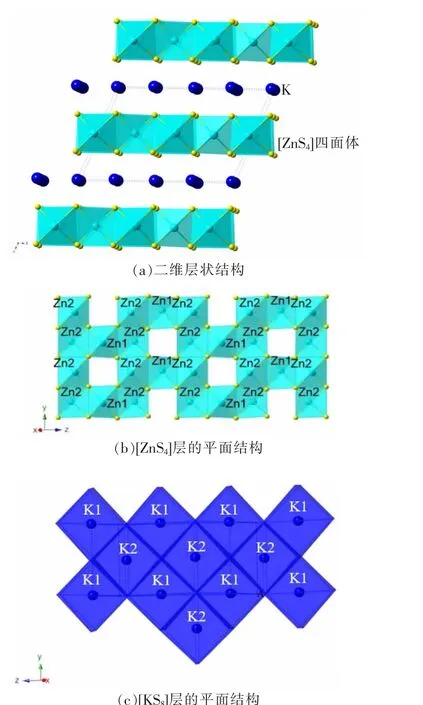
图2 K2Zn3S4 的晶体结构
2.2 成分分析
K2Zn3S4的元素组成和分布采用SEM-EDAX 部件分析,图3 为单晶样品的SEM 二次电子像和元素面扫描图。可见,样品有规则、平滑的表面,对应其单晶形态。 K、Zn 和S 分布均匀,表明样品为单相,没有第二相出现。

图3 K2Zn3S4 的SEM 图及元素分布图
图4 为K2Zn3S44 的EDAX 谱,可以看出,样品中只有K、Zn 和S 三种元素。 测定的原子百分比为K∶Zn∶S=19.33∶29.00∶37.08=2.00∶3.01∶3.84,接近K∶Zn∶S=2∶3∶4 的名义比值。

图4 K2Zn3S4 的EDAX 谱
2.3 拉曼光谱
K2Zn3S4的测量拉曼光谱如图5 所示, 在70、94、212、284 和315 cm-1处存在明显的散射峰。K2Zn3S4的空间群为P2/c(No.13),对应的点群为C2h(2/m),其特征标表见表3。 从表3 可知,振动模Ag和Bg为拉曼活性,而振动模Au和Bu为红外活性。 因P2/c 为中心反演对称性的空间群,拉曼和红外活性振动模是相互排斥的。

表3 C2h 的特征标表
K2Zn3S4的拉曼活性振动模的不可约表示为

利用CASTEP 模块计算得到K2Zn3S4的拉曼光谱, 如图5 所示。 可以看出315 cm-1处的拉曼峰对应振动模Ag,由Zn-S 键的伸展引起的(图6(a))。 284 和212 cm-1处的拉曼峰也都对应振动模Ag,分别表征了Zn-S 键的弯曲和伸展(图6(b)和(c))。 70 cm-1处的拉曼峰表征K-S 和Zn-S 键构成网络的平移(图6(d)),对应振动模Bg。在212、284 和315 cm-1处的散射峰是由Zn-S 键的作用所引起的振动峰。这种拉曼峰也常见于其他硫族化合物,如Li2ZnGeS4(260 cm-1)[23]、Na2ZnGe2S6(294 cm-1)[24]、Cu2ZnGeS4(273 和293 cm-1)[25]和Cu2ZnSnS4(284 和332 cm-1)[26]。 另外,由于K2Zn3S4的吸收峰均在370 cm-1范围以内,暗示着该化合物的红外透光范围可达27 μm。

图5 K2Zn3S4 的拉曼光谱 (竖线为振动模的理论峰位)

图6 K2Zn3S4 的简正模式
2.4 红外光谱
K2Zn3S4的红外光谱如图7 所示,波数的测试范围从600~4 000 cm-1,对应的波长为2.5~16.7 μm。对于K2Zn3S4,波数1 500 cm-1以上透过率在98%以上, 表明了K2Zn3S4对于波长为2.5~6.7 μm 的中远红外光几乎不吸收,具有极好的透过性。 在600~1 500 cm-1之间存在几个吸收峰,但是透过率也在90%以上, 说明K2Zn3S4在6.7~16.7 μm 范围的远红外光具有较高的透过率。

图7 K2Zn3S4 的红外光谱
2.5 漫反射光谱
Kubelka-Munk 公式将漫反射光谱数据转变为吸光度谱[27],如图8 所示。 Kubelka-Munk 公式如下
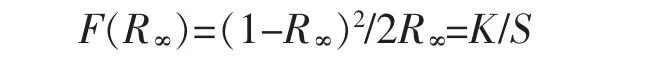
其中,R∞为绝对反射率(把硫酸钡的反射率标定为100%),K 为吸收系数,S 为散射系数。 由图8 知,K2Zn3S4的光学吸收边在370 nm 附近,说明光学带隙约为3.35 eV。
在光学吸收边附近可用Tauc 方程判断半导体性质[28],此方程为
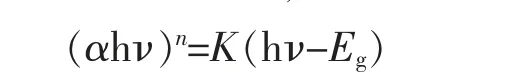
当指数n=2 时,表明样品为直接带隙半导体;当n=1/2 时,表明样品为间接带隙半导体。 利用Tauc 方程对K2Zn3S4的吸光度谱做(F(R)E)2-E 曲线,见图8(b)。 可见,在吸收边附近呈现很好的线性,说明K2Zn3S4具有直接带隙。延长线性关系至(F(R)E)2=0 得到带隙Eg=3.43 eV,属于宽禁带半导体。相较于ZnS 的Eg=3.50 eV[29],K 的引入略微减小了禁带宽度。 K+作为电荷平衡插入离子,主要通过静电力与阴离子骨架相互作用,因此不会显著扰动[Zn3S4]2-的能带结构。

图8 K2Zn3S4 的漫反射光谱(a)和(F(R)E)2 光谱(b)
2.6 能带结构
为了研究K2Zn3S4的晶体结构和光学性质之间的关系,利用第一性原理计算了能带结构,如图9 所示。 可见K2Zn3S4的费米能级(EF)处不存在任何能带,价带和导带之间为禁带,验证了K2Zn3S4的半导体特性。 此外,K2Zn3S4的价带顶和导带底均位于G 点(000),表明该化合物为直接带隙半导体,这与之前的漫反射光谱分析的半导体类型一致。

图9 K2Zn3S4 的能带结构
K2Zn3S4的态密度如图10 所示, 在-15~-10 eV 的低能级区域主要是S-3s 轨道和K-4p 轨道杂化, 反应了K-S 之间的共价键合;-7 到0 eV 的范围,包括价带顶部份,主要由S-3p 和Zn 轨道占据,反映了Zn-S 的共价结合;而导带底部的轨道主要由K、Zn 和S 的轨道贡献。 因此,K2Zn3S4的能带间隙主要由[ZnS4]四面体和[KS8]伪立方体决定。 相对于二元ZnS,K 的引入降低了导带底电子轨道的能量,从而得到了比ZnS 稍低的带隙。

图10 K2Zn3S4 的态密度
3 结语
笔者系统研究了K2Zn3S4的单晶生长、 晶体结构和光学性质,通过助溶剂法生长出单斜的K2Zn3S4单晶,属于中心对称结构。 得到K2Zn3S4的拉曼光谱,并通过计算指定了相应峰对应的振动模。 K2Zn3S4的特征峰在284 和315 cm-1处,对应Zn-S 键弯曲和伸展的振动模Ag。拉曼和红外光谱测量表明K2Zn3S4对波长为2.5~16.7 μm 的中远红外光具有极好的透过性,在红外光学领域内有潜在的应用前景。 通过漫反射光谱实验和第一性原理计算表明K2Zn3S4属于直接带隙半导体,禁带宽度为3.43 eV,表明其在光电和透明导电器件中具有潜在的应用价值。
