金属二硼化物表面性质的密度泛函计算
2021-11-26张桓博何志军
张桓博,王 健,何志军
(辽宁科技大学 理学院,辽宁 鞍山114051)
金属二硼化物(MB2)具有良好的耐磨损、高硬度、高熔点和高电导率等特性,被广泛应用于国防、电子工业、航空航天等领域,是重要的功能结构材料。2001年,熊光成发现MgB2的高温超导性能,其临界温度T0为39 K,这一发现震惊了整个超导材料界,对金属硼化物特性的研究成为热点[1]。2008年,Shein等[2]采用第一原理计算了MgB2(M=Mg、Al、Zr、Nb)非化学计量比条件下的电子结构状态,揭示了金属二硼化物稳定的原子构型与金属阳离子类型有关。2010年,罗晓光等[3]通过第一性原理计算了包括HfB2、TaB2、NbB2、WB2在内的IV-VI族过渡金属二硼化物的电子结构、相稳定性和静/动力学性质,并进行了分子动力学模拟。2011年,喻亮等[4]通过第一原理计算了ZrB2电子结构和光学性质,进一步揭示了ZrB2物理和光学特性。2013年,赵立凯等[5]采用第一原理密度泛函(Density functional theory,DFT)计算了5d过渡金属二硼化物的电子结构和热力学性质,发现从HfB2到AuB2生成焓逐步增加,生成焓的计算结果与化合物热力学稳定性相符。2018年,黄文杰等[6]将VB2以中间合金的形式作为增强相加入到A390合金中,发现A390的性能得到提高。硼化物还可以应用于电子发射领域,LaB6是目前重要的电子发射材料,为了提高其发射性能,比较常用的方法就是通过掺杂进行表面改性。2015年,Taran等[7]研究发现,在LaB6中掺杂MB2形成的复合材料可以改善LaB6的发射特性,降低功函数。Tyson等[8]发现,LaB6/VB2共晶系具有低功函数和高机械强度的特点,是高性能热电子发射体的理想选择。电子发射性质与材料的表面性质密切相关,需要全面了解MB2的表面性质,而目前对于MB2表面性质的研究并不多见。因此,本文采用第一原理密度泛函方法对四种常见MB2(ZrB2、VB2、TaB2、NbB2)的表面性质进行计算与分析,研究其主要密排面(0001)、(10-10)、(11-20)的电子功函数和表面能,探讨电子功函数和表面能的变化规律,建立较为完备的数据库,为MB2表面性质的研究提供理论支持。
1 计算方法与模型
1.1 计算方法
计算采用基于密度泛函理论的从头算量子力学程序包VASP(Vienna ab-initio simulation package)[9]完成,采用周期性边界条件,并使用平面缀加波(Plane additive wave,PAW)赝势[10]描述离子与价电子之间的互相作用。电子交换关联能采用广义梯度近似(Generalized gradient approximation,GGA)方法[11],利用具有周期性结构的片层Slab模型模拟金属表面。平面波的截断能是500 eV。不考虑自旋,Slab模型之间的真空层厚度为1.5 nm,忽略相邻表面之间的相互作用。Slab模型上下表面充分弛豫,当原子间力的最大值小于0.02 eV时弛豫结束。
1.2 计算模型
MB2(M=Zr、V、Ta、Nb)的晶体结构为简单六方晶体,硼原子和金属原子呈交错排列,空间组为P6/mmm,详见图1。为了更详尽地描述其表面性质,本文截取3个典型密排面,即(0001)、(10-10)、(11-20)面。由于截取的位置不同,终结面原子构成也不同,分别为:只含金属原子表面、只含硼原子表面及由金属和硼原子组成的混合面。如(0001)表面,如果从虚线处截取,形成的两个表面中,一个是B原子为终结面,另一个是M原子为终结面。终结面的原子不同,其表面性质就不一样。因此,本文构建了不同原子终结面的Slab模型,如图2所示。其中图2a和图2b为(0001)面分别以B原子为终结面B-Term(0001)和M原子为终结面M-Term(0001)的Slab模型。(10-10)面的终结面有三种情况,其中图2c为M原子终结面M-Term(10-10),图2d和图2e都是以B原子为终结面的两种不同的结构,分别为B1-Term(10-10)和B2-Term(10-10)。(11-20)面只有一种终结面,为M和B原子的混合面M,B-Term(11-20)。
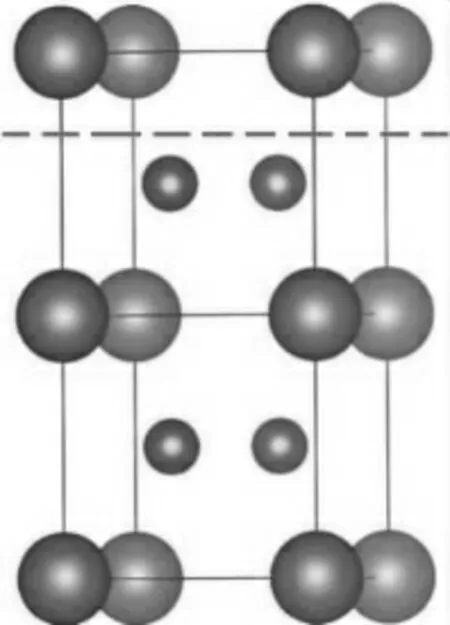
图1 MB2的三维模型(1×1×2)Fig.1 Three dimensional model of MB2(1×1×2)
1.3 电子功函数
电子功函数Φ的定义是把一个电子从物体内部的费米能级Ef移到物体表面所需要的最小功。根据这个定义,对于Slab模型,可表示为[12]
式中:VVAC代表真空中的静电能,V;Ef是系统的费米能级,V。
1.4 表面能
表面能是判断表面稳定性的一个重要物理参数。表面能通常表示为单位面积的吉布斯自由能[13]。一个无限的晶体沿某一晶面劈开,生成两个半无限的子系统,各自均构成一个面积为S的表面。而表面能便是劈开过程中所需要的功。对于MB2对称的片层结构,其化学组分一般是非化学计量比,不能直接求解其表面能。本文采用非化学计量比表面能计算方法[14]。
以图2中MB2(0001)面的两个Slab模型为例,其表面只有单一的M或B原子。为了得到每一种终结面的表面能,在计算时使用非化学计量比的片层结构。考虑到表面的压强P和温度T,MB2(0001)面的表面能σMB2计算式[15]
式中:Asur是表面积;Eslab是完全弛豫的M终结面或B终结面表面的总能量;NM和NB分别是晶体中M和B原子的数量分别是片层中M和B的化学势;PV和TS分别表示在一定状态下,表面能受压强和温度影响的值。0 K时PV和TS项可以忽略不计。
块体MB2中,M和B原子的化学势之和与同体材料MB2的化学势始终保持平衡。因此,块体MB2的化学势可以表示为
又因为块体MB2化学势定义
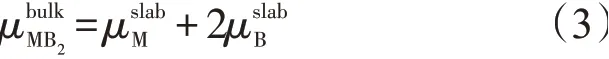
结合式(3)和式(2),得到表面能计算式
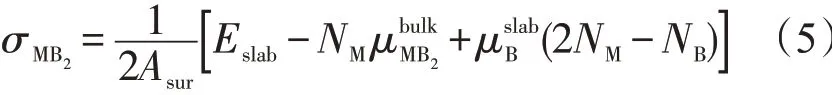
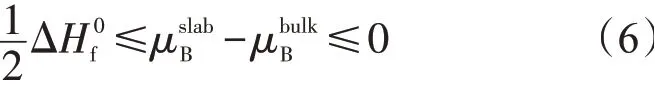
M端和B端终结面的表面能是B化学势的函数,根据式(5)和式(6)分别计算出4种MB2在(0001)、(10-10)、(11-20)面M端和B端终结面表面能的取值范围。
2 结果与讨论
在建结构模型时,为得到与实验中更匹配的晶胞结构,进而获得更复杂的结构或预测其理化性质,需要对模型进行结构优化。本文对四种MB2单胞的晶格常数a和c进行优化计算。晶格常数就是晶胞的边长,也就是四种MB2单胞的三个轴长。优化后得到的晶格常数a和c数值变化通常不明显,一般采用轴比a/c体现晶格常数变化大小。晶格常数计算结果详见表1。与文献中实验值进行比较,误差在3%以内,计算值与实验值符合较好,说明本文采用的计算方法合理。
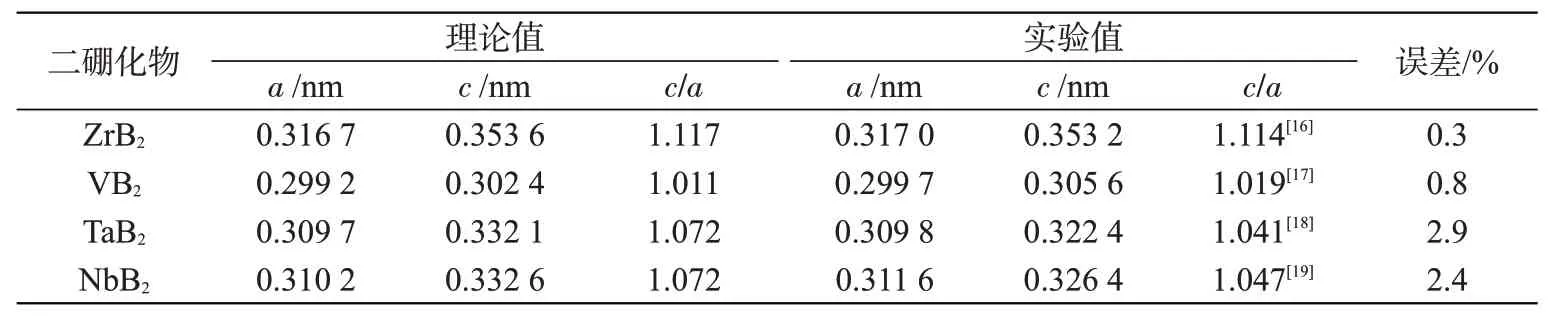
表1 四种金属二硼化物结构优化后的晶格参数Tab.1 Lattice parameters of four structure-optimized metal diborides
2.1 电子功函数
电子功函数是研究物质表面性能最重要的物理参数之一,不仅能反映硼化物表面特性,例如表面偶极子分布和表面电子的分布情况等,也可以反映材料表面活性,即表面获得和失去电子的能力。在电子发射研究中,发射电流密度与材料功函数和所加电场强度密切相关。因此,电子功函数也是衡量电子发射材料发射性能优劣的重要指标。
以ZrB2(0001)面Zr终结面与B终结面为例,电子功函数计算结果如图3所示。图3中水平虚线代表费米能级,曲线周期震荡的部分代表势能曲线在原子层间的变化,右侧的平台代表真空能级,真空能级和费米能级的差即为电子功函数Φ。不同原子终结面的电子功函数不同。
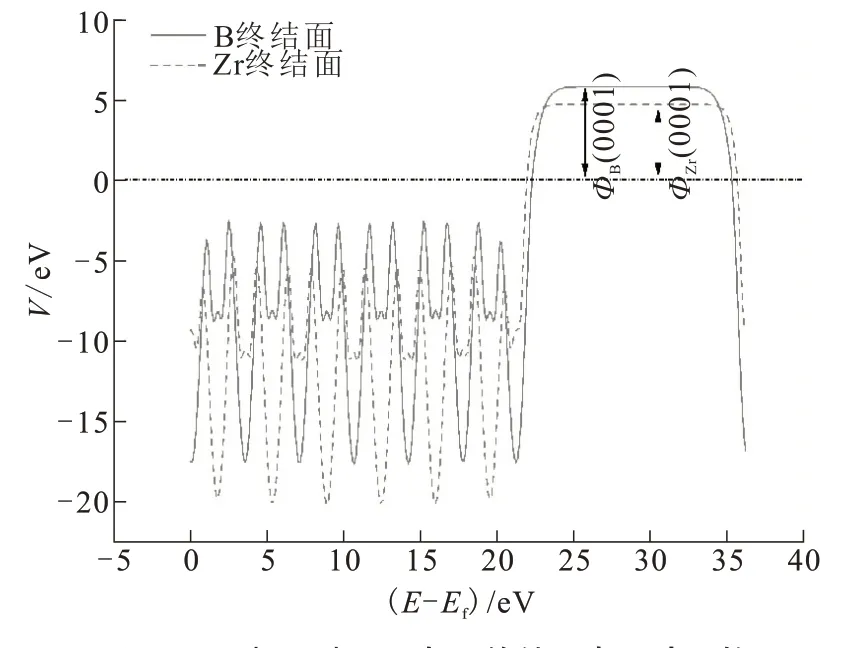
图3 ZrB2(0001)面Zr与B终结面电子功函数Fig.3 Electronic work function of Zr and B terminals of ZrB2(0001)plane
采用同样方法计算出其他MB2各个面的电子功函数,详见表2。四种MB2计算值与已有的实验值进行比较,符合较好。以M为终结面的功函数低于以B为终结面的电子功函数,这可能与元素的电负性和活泼性紧密相关。四种MB2金属面的电子功函数遵从相同的变化规律:Φ(10-10)<Φ(0001)<Φ(11-20)。其中,ZrB2(10-10)金属面的电子功函数最低,为3.757 eV。
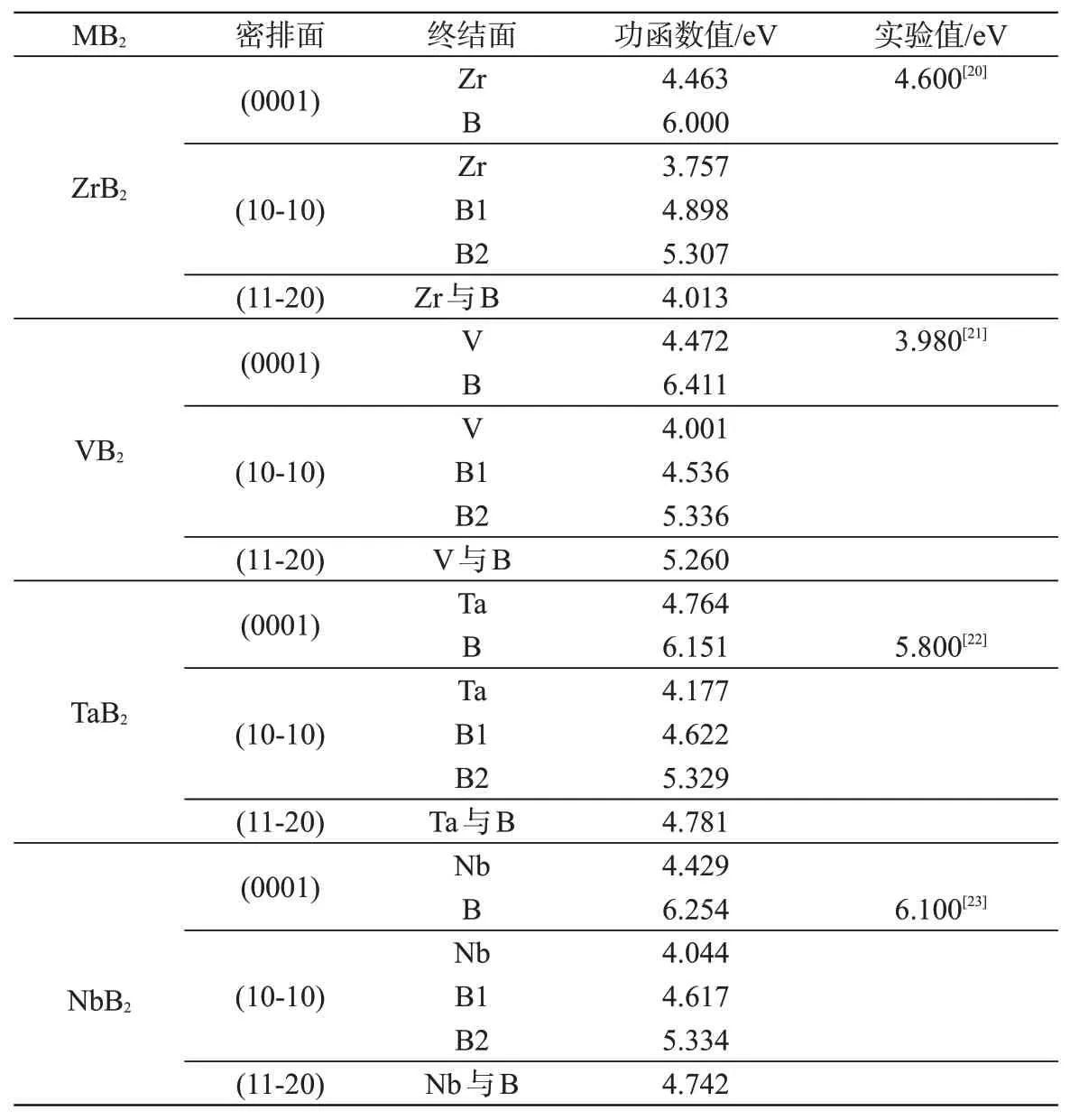
表2 MB2的主要密排面的电子功函数Tab.2 Electronic work function of MB2
2.2 表面能
根据式(4)分别作出四种MB2的(0001)、(10-10)、(11-20)面M和B终结面的表面能变化曲线,如图4所示。
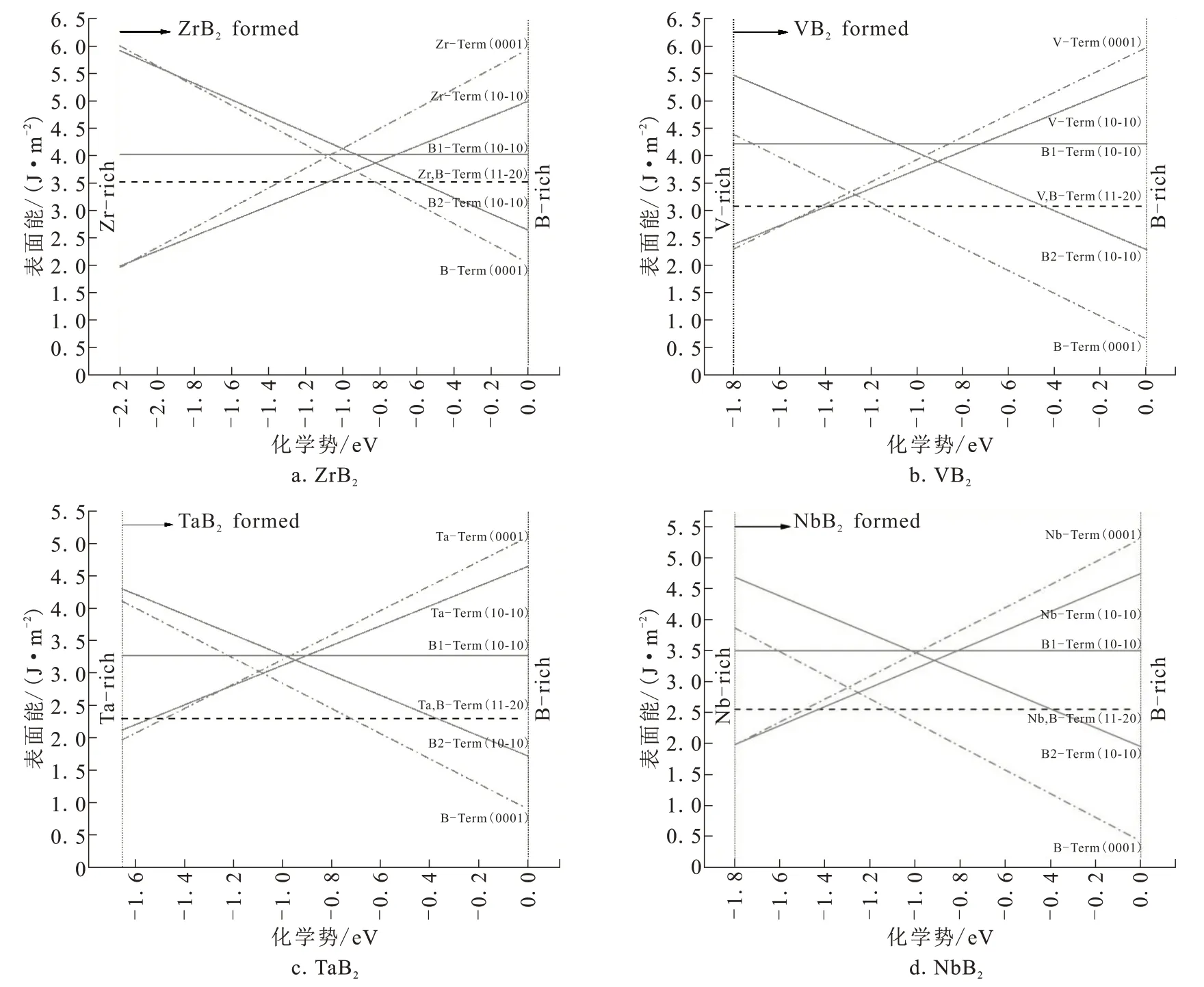
图4 四种MB2的(0001)、(10-10)、(11-20)面上B端和M端表面能和化学势的关系Fig.4 Relationship between surface energy and chemical potential of B-terminal and M-terminal on four MB2 planes((0001),(10-10),(11-20))
对于ZrB2,随着B化学势的升高,Zr原子终结面Zr-Term(0001)表面能线性增大,B原子终结面B-Term(0001)表面能线性减小。在化学势变化的大部分区域内,Zr-Term(0001)表面能比B-Term(0001)表面能低,表明此范围中Zr-Term(0001)面更加稳定[24]。对于(10-10)面,在化学势变化的绝大部分的区域内(-2.2~0.779 eV),Zr-Term(10-10)面比B2-Term(10-10)表面能低,说明Zr-Term(10-10)面稳定性更高。此结果与文献[22,24-26]的结论一致。B1-Term(10-10)面和(11-20)面为等化学配比面,表面能为定值,分别为4.071 J/m2和3.512 J/m2。
Ⅴ族元素V、Ta、Nb的MB2在(0001)、(10-10)、(11-20)面的表面能与化学势的函数关系规律一致,即在B化学势较低的范围内,M终结面表面能相对较低,较为稳定;在B化学势高的范围内,B终结面的表面能比较低,更加稳定。从整体上看,B终结面表面能在化学势更大范围内比M终结面的表面能低。因此,VB2、TaB2、NbB2三种MB2均以B原子为终结面的结构更稳定,这与文献[24,27-29]一致。
3 结论
本文基于第一原理密度泛函理论,采用VASP软件分别计算了ZrB2、VB2、TaB2、NbB2四种MB2的(0001)、(10-10)、(11-20)面的表面能和电子功函数,建立了较为详细的数据库。计算结果表明,四种MB2各个面的功函数值均为金属终结面更低。ZrB2的Zr终结面较B终结面表面能更低,更为稳定。而三种Ⅴ族元素的MB2则以B终结面表面能更低,同族元素呈现相似的表面能变化规律。
