间歇性缺氧对睡眠呼吸暂停大鼠模型肠道菌群结构的影响*
2021-11-24张珍连成林杰艾孜买提肉孜江姚巧玲
张珍连 成林杰 艾孜买提·肉孜江 姚巧玲
(新疆医科大学基础医学院生理学教研室,新疆 乌鲁木齐 830000)
阻塞性睡眠呼吸暂停综合征(obstructive sleep apnea syndrom, OSAS)是以夜间间歇性缺氧(Intermittent hypoxia, IH)、睡眠片段化为主要病理特征的常见呼吸系统疾病。成人中49.7%的男性和23.4%的女性有中至重度OSAS[1]。且OSAS可显著增加认知障碍、肥胖、心血管疾病、癌症等发病风险[2-8]。因此,OSAS已成为备受关注的公共卫生问题。目前多数学者认为IH是OSAS引起多器官损害的主要原因。肠道菌群在维持全身稳态方面起着至关重要的作用。肠道菌群失调与肥胖、糖尿病、神经退行性疾病、心血管疾病密切相关[9-14]。Moreno等[15]在OSAS小鼠模型中发现IH可导致肠道微生物组内的低氧/复氧循环事件,从而改变肠道菌群组成和生物多样性。提示IH可能通过改变肠道菌群参与OSAS及其相关多器官病变。然而目前关于IH对肠道菌群影响的研究报道尚少,因此本研究构建了IH大鼠模型,并首次报道大鼠在IH条件下肠道菌群结构变化特征,旨在为研究OSAS及其多器官病变的发病机制提供科学依据。
1 资料与方法
1.1 研究对象 选用16只6周龄清洁级雄性SD大鼠,体重250g左右,来自新疆医科大学动物实验中心)作为研究对象,开始前10天及为期4周的研究期间,动物被安置在新疆医科大学动物中心由温度和灯光控制的房间,饲养在标准笼子里,接受自来水和消毒的标准食品。本研究对大鼠的处置符合动物伦理要求,并得到了新疆医科大学动物研究伦理委员会的批准。
1.2 实验设计 16只大鼠随机分为常氧组(NC组)及间歇性缺氧组(IH组),每组8只。其中IH组在大鼠睡眠时相占主导的白天(9am-9pm)给予持续性IH刺激(小动物呼吸机:上海玉研科学仪器有限公司,上海,IOplus),IH参数采用我们以往的实验进行[16],即氧分压在6%~21%范围内循环,每分钟一个循环,一小时60个循环,持续给予4周IH刺激;常氧组实验期间维持常氧分压(21%)。
1.3 样品采集 在4周间歇性缺氧或常氧处理实验结束后,分别采集每只大鼠粪便并放入提前标记好的1.5 mL 无菌离心管中,-80 ℃保存。
1.4 DNA 提取和16S rRNA 高通量测序 使用E.Z.N.A.©DNA提取试剂盒(Omega Bio-tek,USA)提取试剂盒提取大鼠粪便中总DNA,并使用NanoDrop 2000©分光光度计(Thermo Fisher Scientific,USA,NanoDrop2000)对DNA进行定量检测。对细菌的16S rDNA 基因V3-V4 高突变片段设计通用引物,测序区域为338F-06R,引物序列为338F(ACTCCTACGGGAGGCAGCAG)和806R(GGACTACHVGGGTWTCTAAT)。PCR 反应体系为20 μL,包括10 μL 2 ×PCR Master Mix Solution、5 μM 正反引物、10 ng模板DNA。反应程序为95 ℃ 3 min;95 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s,27 个循环;72 ℃ 10 min。使用2% 琼脂糖凝胶(Biowest, Espana)电泳检测PCR 扩增产物,利用AxyPrep DNA凝胶回收试剂盒(Axygen Biosciences,USA)对目的片段进行切胶回收。利用微型荧光计(Promega,USA, QuantusTMFluorometer)对纯化后的片段进行定量检测。根据定量结果对各样本进行相应比例混合,利用测序仪(Illumina, San Diego, USA, Illumina MiSeq)进行测序。
1.5 生物信息学分析 数据分析使用美吉生物云平台的免费在线平台(www.i-sanger.com)。首先根据PE reads之间的overlap关系,将成对的reads拼接(merge)成一条序列,同时对reads的质量和merge的效果进行质控过滤,根据序列首尾两端的barcode和引物序列区分样品得到有效序列,并校正序列方向,即为优化数据。采用fastp、FLASH软件进行序列去杂。采用Usearch进行OTU聚类分析:对优化序列提取非重复序列;去除没有重复的单序列;按照97%相似性对非重复序列(不含单序列)进行OTU聚类,在聚类过程中去除嵌合体,得到OTU的代表序列;将所有优化序列map至OTU代表序列,选出与代表序列相似性在97%以上的序列,生成OTU表格。采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,并分别在各个分类水平:域、界、门、纲、目、科、属、种统计各样本的群落物种组成。根据OTU分析结果,利用Shannon,Chao 1,coverage指数对样本进行α多样性分析,反映微生物群落的丰富度和多样性及群落覆盖度。采用物种Venn图和群落组成分析两组样本中所共有和独有的物种(如OUT)数目及不同分组在各分类水平(如域、界、门、纲、目、科、属、种、OTU等)上的物种组成情况。采用PCoA分析研究样本群落组成的相似性或差异性。采用Lefse多级物种差异判别分析进一步对两组间肠道菌群物种丰度差异进行分析。采用共线性网络及单因素网络分析样本与物种间的共存关系。
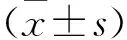
2 结果
2.1 IH组和NC组肠道菌群多样性分析 IH组和NC组共有424544个16S rRNA基因序列,平均每个样本有26534个序列通过了QIIME的筛选。IH组OTUs 数量(1044个)与NC组(1078个)相比无显著差异(P>0.05)。IH组与NC组有效OTUs 共1153个,共有OTUs 969个,独有OTUs分别为75、109个。两组的群落多样性(Shannon指数)和群落丰富度(Chao1指数)分析结果显示,间歇性缺氧暴露并未显著增加大鼠粪便样本的群落多样性和丰富度(P>0.05)。IH组和NC组肠道菌群Coverage 指数均接近1,且两组间无显著性差异(P>0.05),提示各样品文库的覆盖率较高,测序结果可以代表了样品中微生物的真实情况,见表1。

表1 IH组和NC组肠道菌群α多样性指数
2.2 IH组和NC组主要肠道菌群组成分析 基于非加权的PCoA 分析结果显示,IH组和NC组的所有样本在坐标组合中分离分布,两组主成分占总变异的28.11%(PC1)和12.96%(PC2)。ANOSIM检验结果证实了组间的显著分离,表明IH组和NC组之间的微生物组成存在显著性差异(P<0.01),见图1。本研究在不同分类学水平上分析了IH组和NC组肠道菌群物种丰度,结果显示,两组大鼠间肠道菌群组成均存在差异。在门水平上,优势菌群依次为厚壁菌门、拟杆菌门、变形杆菌门、软壁菌门。其余的细菌属于所有样本中相对丰度小于1%的其他9个门,见图2A。在科水平上,两组大鼠肠道菌群共计发现52个科,其中包括12个优势菌科,依次为,S24-7,毛螺菌科,瘤胃菌科,普雷沃氏菌科,乳杆菌科,消化链球菌科,脱硫弧菌科,韦荣球菌科,拟杆菌科,柔膜细菌RF9,理研菌科,克里斯滕森菌科。其余的细菌属于所有样本中相对丰度小于1%的其他40个科,见图2B。
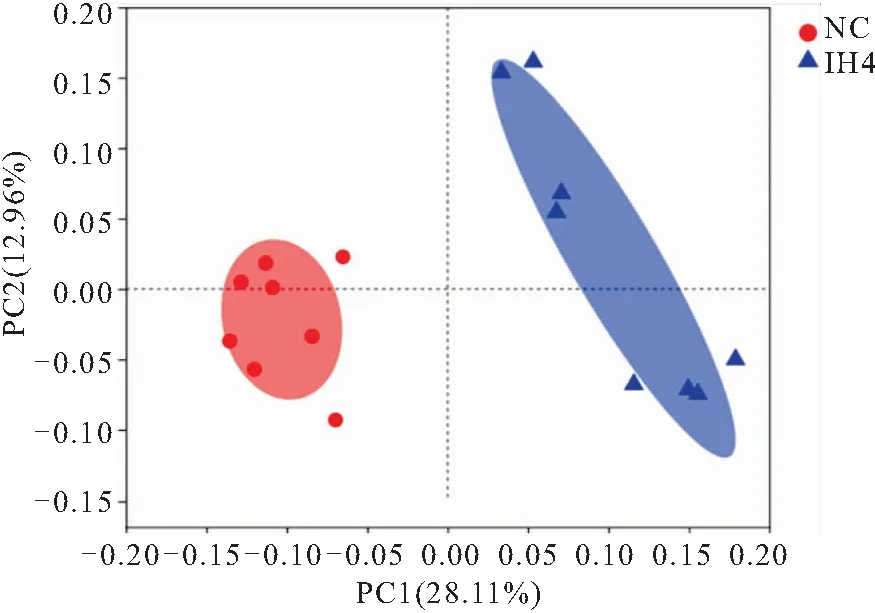
图1 IH组和NC组肠道菌群PCoA分析图
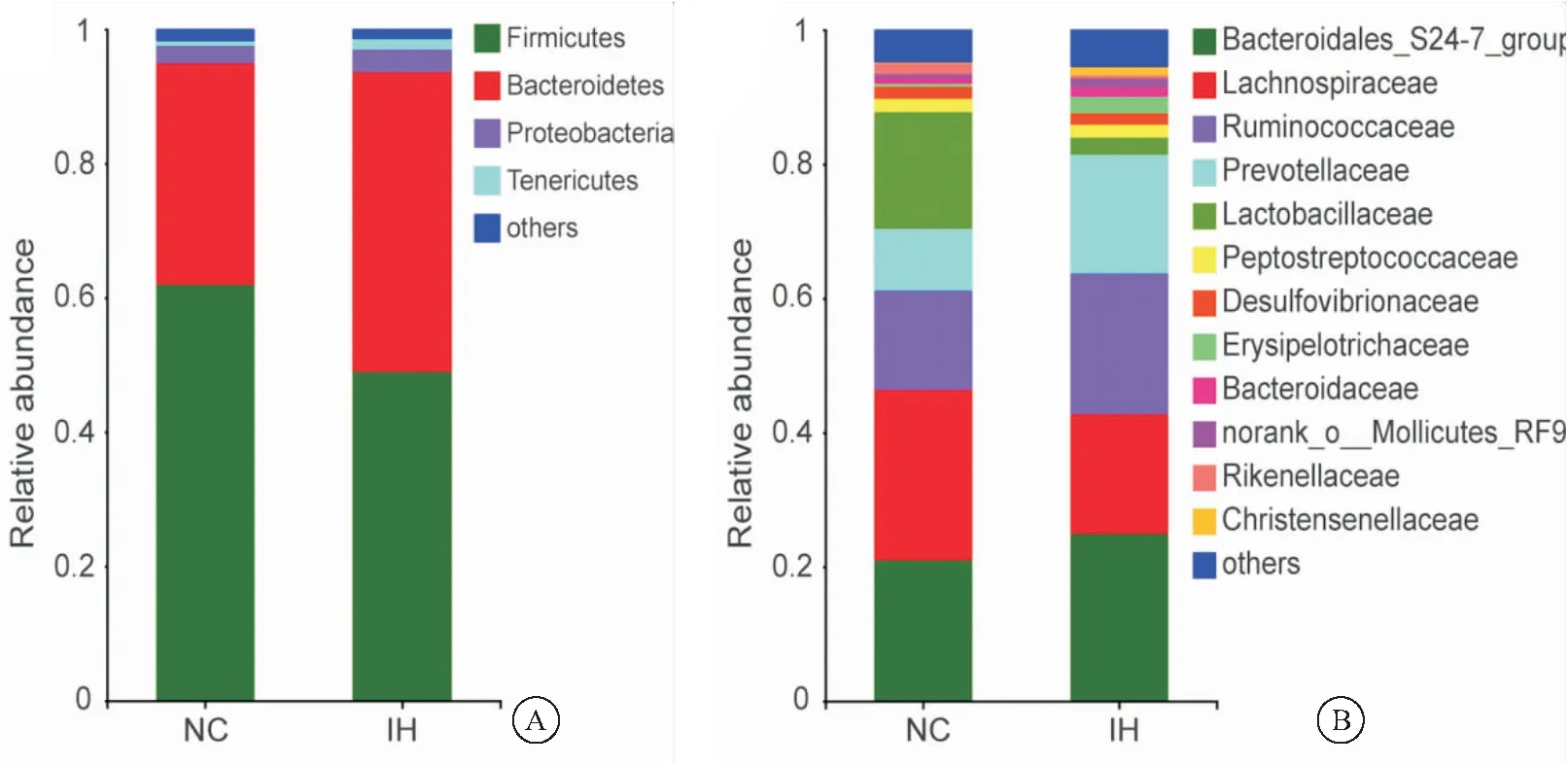
图2 IH组和NC组肠道菌群在门和科分类水平上的相对丰度
2.3 IH组和NC组肠道菌群物种差异分析 Lefse多级物种差异判别分析结果显示,在门水平,拟杆菌门(P<0.05)、软壁菌门(P<0.01)在IH大鼠肠道中显著富集,而厚壁菌门(P<0.05)在NC组大鼠肠道中显著富集,但并未发现间歇性缺氧对大鼠肠道内变形菌门(P>0.05)物种丰度的影响。其余的9个弱势菌门的物种丰度在两组间均无统计学差异(P>0.05)。在科水平上,12个优势菌科中,普雷沃氏菌科(P<0.05),克里斯滕森菌科(P<0.001),韦荣球菌科(P<0.05),柔膜细菌RF9(P<0.01)在IH大鼠肠道中显著富集,理研菌科(P<0.001),乳杆菌科(P<0.01)在NC组大鼠肠道中显著富集。而在弱势菌科中,产碱菌科(IH组:0.624%,NC组:0.075%;P<0.01),红螺菌科(IH组:0.110%,NC组:0.035%;P<0.05),vadinBE97(IH组:0.003%,NC组:0%;P<0.05)IH组显著富集,未发现其他弱势菌科在两组间分布的显著差异(P>0.05,见图3A、图3B。采用共现性网络分析及单因素相关性网络分析结果显示,IH组和NC组间有广泛的物种之间的相互作用。此外,我们在两组均发现了游离的OTUs,IH组和NC组各发现43个、39个游离的OTU样本(图4)。对分类水平总丰度前30的OTU进行单因素相关性网络分析,结果显示56.7% OTUs(17/30)属于厚壁菌门,36.7% OTUs(11/30)属于拟杆菌门,剩余6.6% OTUs(2/30)属于变形菌门(图5)。属于拟杆菌门的OTU928可以与14种主要的OTUs相互作用,属于厚壁菌门的OTU397和OTU405分别可以与14、13种主要的OTUs相互作用,属于拟杆菌门的OTU1011、OTU914和OTU261分别可以与13、13、12种主要的OTUs相互作用,其他两个厚壁菌门(OTU887、OTU592)分别可以与11、10种主要的OTUs相互作用,而拟杆菌门的OTU1073也发现可以11种主要的OTUs相互作用。
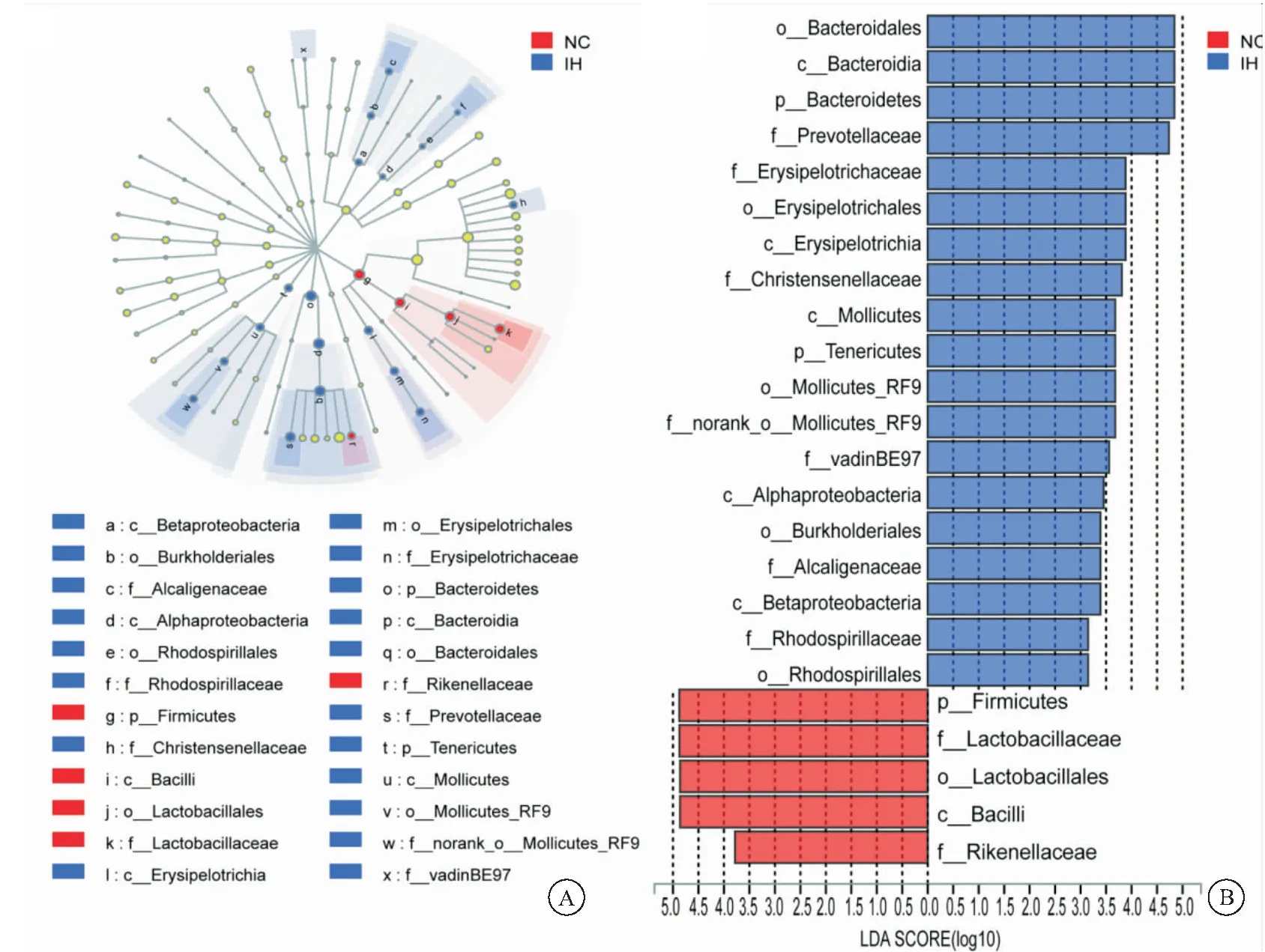
图3 IH组和NC组肠道菌群LEfSe分析图
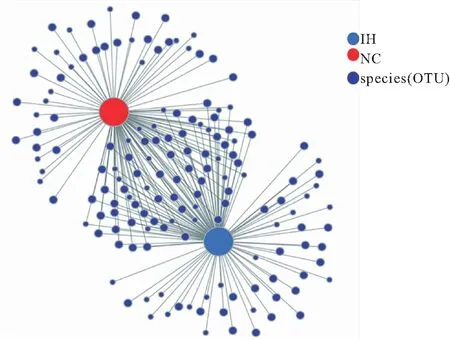
图4 IH组和NC组肠道菌群共线性网络图
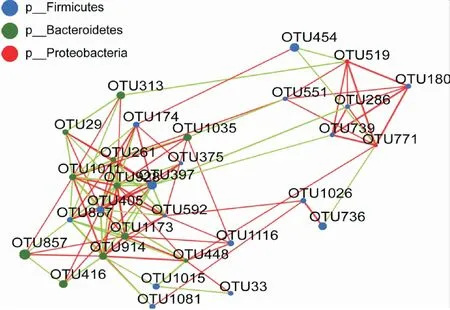
图5 分类水平总丰度前30的OTU单因素相关性网络图
3 讨论
OSAS是一种伴有心血管疾病、代谢异常的全身性、综合性疾病。IH在OSAS及其相关多器官病变中发挥着重要作用,但其作用机制尚不完全明确。研究显示肠道菌群可能与OSAS及其相关的包括新陈代谢在内的多种疾病状态密切相关。因此本研究通过IH模拟OSAS大鼠模型,发现IH可以显著改变OSAS大鼠肠道菌群结构组成,但对菌群的多样性和丰富度并无显著影响。
Moreno-Indias等[15]报道与常氧组小鼠相比,间歇性缺氧处理可显著增加小鼠肠道菌群OTU数量,同时显著上调菌群平均多样性和群落丰富度。与之相反,本研究在大鼠模型上并未发现IH组(1044个)与NC组(1078个)之间OTU数量之间的显著差异。同时两组的群落多样性(Shannon指数)和群落丰富度(Chao1指数)分析结果并未显示间歇性缺氧暴露显著增加大鼠粪便样本的细菌多样性和丰富度。有意思的是,Chih-Yuan Ko等[17]研究了我国93例OSAHS患者,结果发现,相对于对照组,OSAHS患者Chao丰富度、Shannon多样性和Simpson多样性并没有显著增加,该结果与本研究结果类似。对IH组与NC组大鼠肠道菌群物种组成及差异分析结果显示,在门水平两组大鼠间肠道菌群组成存在显著差异。但Moreno-Indias 等人在比较间歇性缺氧组和常氧组时,并未发现小鼠肠道菌群在门水平上的显著差异[15]。此外Chih-Yuan Ko等[17]在OSAHS患者中亦未发现肠道菌群在门水平上与对照组人群之间的显著差异。但在科水平,本研究与Moreno-Indias I等及Chih-Yuan Ko等研究结果类似,均发现优势菌科水平上存在显著差异[15,17]。
本研究发现,优势菌群中,普雷沃氏菌科,克里斯滕森菌科,韦荣球菌科,柔膜细菌RF9在IH大鼠肠道中显著富集,而理研菌科,乳杆菌科在NC组大鼠肠道中显著富集。而弱势菌群中,产碱菌科,红螺菌科,vadinBE97在IH组显著富集。前期研究已报道,间歇性缺氧处理小鼠与正常小鼠相比,肠道内氧含量的总体减少[15],提示IH处理将赋予厌氧菌群生存优势,而兼性厌氧菌甚至好氧菌则处于不利地位,从而引起优势菌群分布变化,尤其是引起专性厌氧菌在间歇性缺氧组中相对富集。
研究发现OSAS患者肠屏障标志物D-乳酸(D-LA)和肠脂肪酸结合蛋白(I-FABP)水平明显升高,提示患者肠道通透性显著增加[18-19]。且动物实验同样发现随着间歇低氧时间的延长,肠道黏膜受损严重,通透性增高,该研究已报道肠道菌群移位可加重对肠系膜淋巴结结构损伤,进而增加氧化应激水平[20]。普雷沃氏菌科已报道参与粘蛋白的降解过程,进而导致肠道通透性改变[21]。本研究结果显示,普雷沃氏菌科在IH组大鼠肠道内显著富集,提示IH可能通过增加普雷沃氏菌科丰度进而引起肠道通透性改变。此外,有研究显示韦荣球菌科为新的伴有代谢综合征肥胖的生物标志物[22],与Chih-Yuan Ko等[17]研究结果相似,本研究亦发现韦荣球菌科在IH组中富集,可能与IH 引起的肥胖密切相关。而克里斯滕森菌科被报道在低体质指数(low body mass index,BMI)的人群中显著富集,且在无菌小鼠中移植里斯滕森菌科后可引起体重显著下降[23]。但本研究发现IH组中里斯滕森菌科显著富集,这与IH诱导肥胖结论并不一致,该结果还需要进一步研究确认。与Moreno-Indias 等及Chih-Yuan Ko等研究结果相反,我们的研究结果显示IH可引起乳杆菌科的显著下调。而肠道中乳酸杆菌能够通过代谢谷氨酸产生中枢神经系统中主要的抑制性神经递质γ-氨基丁酸(GABA)。乳酸杆菌的减少,会影响肠道中GABA的产生,进而导致中枢中GABA的减少。而GABA能系统的功能障碍可能导致认知功能障碍[24-27]。提示IH引起乳杆菌科显著下调可能参与认知功能障碍发生。
4 结论
本研究利用IH大鼠模型,进一步证实间歇性缺氧可引起肠道菌群结构的显著改变,从而改变宿主与肠道菌群之间的稳态关系。本研究为肠道菌群参与OSAS及其多器官病变的发病机制提供了科学依据,而OSAS如何通过肠道菌群调节宿主代谢将值得进一步研究。
