ReaxFF MD局部区域反应追踪与物理性质可视化分析
2021-11-22唐钰杰郑默任春醒李晓霞郭力
唐钰杰,郑默,任春醒,李晓霞,*,郭力
1中国科学院过程工程研究所多相复杂系统国家重点实验室,北京 100190
2中国科学院大学化学工程学院,北京 100049
3中国科学院绿色制造创新研究院,北京 100190
关键字:ReaxFF MD;区域反应追踪;物理性质;可视化
1 引言
反应分子动力学是将反应力场 ReaxFF(Reactive Force Field)和分子动力学MD (Molecular Dynamics)结合的分子模拟方法。基于Adri van Duin等1提出的反应力场ReaxFF,ReaxFF MD可较好地模拟复杂分子反应体系中键的生成和断裂随时间的变化,从而模拟化学反应随时间的演化。目前,国际上主流的分子模拟方法包括分子动力学和量子力学方法。经典分子动力学方法基于牛顿力学主要描述体系的物理性质,可模拟大规模分子体系(~100000 – ~1000000原子)随时间的动态演化;但由于原子的连接关系和电荷保持不变,无法描述化学反应。量子力学基于薛定谔方程,可精确描述体系中电子运动状态,从而描述可能的反应路径;但其计算代价高昂,能够应用体系的原子规模相对较小(~100原子)。为此,基于量子力学的分子动力学模拟可考察体系键生成和断裂的动态过程,但因计算能力的限制,难以应用于复杂分子体系。ReaxFF MD方法基于键级描述力势中所有与成断键相关的能量项,包括键长、键角、扭转二面角、过配位和配位不足校正、氢键作用、其他相互作用的校正如孤对电子能、三体共轭、四体共轭等;采用基于Taper校正的修正Morse势描述范德华非键作用;采用原子点电荷描述库仑静电作用,并且利用电负性平衡算法动态更新每个分子动力学时间步的原子电荷。由于无需预设反应路径,可平滑描述多分子体系化学反应随时间的演化过程,因此,ReaxFF MD方法是一种有潜力模拟和揭示复杂分子体系化学反应机理的新方法。
分析ReaxFF MD模拟结果中所蕴含的化学反应信息,对利用ReaxFF MD模拟方法认识复杂体系的反应机理极为关键。目前,国际上集成了ReaxFF MD模拟计算程序的主流平台较多,有LAMMPS2、AMS (原名是ADF)3、Materials Studio(MS),但专门分析ReaxFF MD模拟结果中化学反应的程序系统仍然十分缺乏。例如,MS中GULP模块的ReaxFF 6.04虽然可以进行反应体系的模拟,但并未提供专门针对ReaxFF MD化学反应信息的分析工具,其自带的工具主要用于分析经典分子动力学模拟的物理过程,只能观察ReaxFF MD模拟得到的分子体系轨迹变化,不能获得化学反应信息。从发表的ReaxFF MD模拟应用文章可知,ReaxFF MD模拟结果化学反应工具主要为LAMMPS平台开源的reax/c模块5,其反应信息分析结果为基于分子式的分子数量随时间的演化,既不能直接给出反应细节(特别是反应位点的信息),也不能区分同分异构体。
随着ReaxFF力场和模拟应用的快速发展,在商业化的分子动力学模拟平台中集成和发展专门用于分析ReaxFF MD模拟结果的程序在最近5年内得到重视。AMS是最早集成LAMMPS中reax/c的化学反应分析功能的平台,同样仅提供基于分子式的分子数量统计,仅通过提供图形化的数据提升了分析结果的直观性;其最近两年的版本集成了德国亚琛大学Döntgen等6发布的一个反应分析程序,该程序建立在采用270个原子研究甲烷氧化基元反应的基础之上,主要进步是可以计算基元反应的反应速率,但难以应用于~10000原子规模的复杂分子体系。MAPS平台的REAXFF ANALYSIS插件则提供了支持LAMMPS中reax/c的模拟结果表格化显示的功能,可识别分子、物种、反应以及计算分子寿命7。由此可见,商业化的分子动力学模拟平台所集成的反应分析程序主要基于开源程序或个别研究者的小程序,在反应分析核心能力的进一步发展上仍然欠缺。最近两年,在ReaxFF MD模拟的反应网络自动生成方面取得了进展。华东师范大学朱通等构建的反应网络自动生成程序ReacNetGenerator采用了隐马尔科夫链方法平滑震荡的基元反应是一个新的尝试,可显著提升生成合理反应网络的效率,已应用于甲烷燃烧的机理研究和4组分RP-3替代燃料氧化的反应网络生成8。由于要预先手动标注一部分噪声反应用于隐马尔科夫链相关矩阵的计算,应用于复杂燃料氧化反应网络的自动生成还有待更多的工作。上海交通大学吴量和孙淮等9则发展了基于优化的键级截断值计算ReaxFF MD模拟的基元反应速率常数、再结合直接关系图法对反应进行机理简化的方法,将其应用于氢气燃烧的ReaxFF MD模拟结果分析,获得了合理的简化动力学模型,展现了从ReaxFF MD模拟基元反应直接获得动力学性质的潜力。总体而言,ReaxFF MD模拟的反应机理分析方法取得了明显进展。但与快速发展的ReaxFF MD模拟应用相比,专门用于分析ReaxFF MD模拟结果的方法和程序实现策略仍然滞后,特别是针对大规模复杂体系的ReaxFF MD模拟应用的化学反应分析方法仍然是挑战。
作者课题组近年来面向国家能源利用相关的重大需求,致力于发展大规模ReaxFF MD模拟的方法,建立了国际首个基于GPU并行ReaxFF MD模拟程序系统GMD-Reax,显著提升了10000–100000原子规模在单GPU计算节点上模拟计算的效率10;基于化学信息学方法建立了ReaxFF MD反应分析与可视化程序系统VARxMD (Visualization and Analysis of ReaxFF Molecular Dynamics)11,12。相比于国际上已有的ReaxFF MD的反应分析工具,VARxMD在处理大规模分子体系模拟结果方面具有领先优势和独特性,其反应分析功能建立在3D化学结构对唯一物种识别、物种反应位点识别、键类型识别的基础之上,可基于相邻时刻之间的成断键信息自动生成完整的化学反应列表,并进行反应位点的2D和3D结构可视化显示;再者,基于反应物或产物的化学结构、官能团和反应位点的检索,可进一步对反应路径进行分类,并图形化展示反应路径的演化13。VARxMD的最新发展是特定反应物和特定生成物之间反应网络的自动生成与可视化14。VARxMD已经在大规模ReaxFF MD模拟高温热解和氧化反应过程、揭示其复杂的反应机理应用中发挥了独特作用15–18。
在应用于复杂过程模拟的反应机理分析时,目前的VARxMD仍存在一定的局限。首先,目前的VARxMD主要用于分析ReaxFF MD模拟体系所蕴含的化学反应信息,并不包含在经典分子动力学模拟中常用的物理过程参数分析,而一些真实的复杂反应过程是物理和化学变化同时伴随发生。例如煤在高温热解过程中,多个大片段焦油自由基分子会重聚形成更大片段的煤焦前驱体分子。不断变化的煤焦前驱体分子夹杂在煤热解体系中,要对其结构特征和时空性质进行表征分析存在困难;难以考察煤热解反应体系中两个煤颗粒之间的相互作用;轻焦油从煤颗粒微孔的逸出与微孔的大小、分布的关系也难以获得。此外,目前VARxMD的反应分析功能主要着眼于模拟体系的全局反应,已有的物种检索功能和3D化学结构拾取功能可以聚焦于特定的反应物或生成物分子,尚不能聚焦于模拟体系的特定反应区域,但局部区域的反应追踪与分析对一些特定的过程极为重要。例如含能材料在外部刺激条件下会形成局部升温并可能演变为爆燃爆轰过程的触发,考察特定热点区域的物理和化学变化是研究者极为关注的问题;又如模拟分子筛催化反应体系时,研究人员关心反应物种在分子筛催化剂表面的物理吸附和化学反应细节。然而到目前为止,国内外的ReaxFF MD反应分析程序系统尚未聚焦于复杂体系局部区域的化学反应分析,难以回溯和追踪特定区域的化学反应。这一重要分析能力的缺失,限制了ReaxFF MD应用于更多复杂过程的反应机理探索。
为此,我们基于在ReaxFF MD模拟反应分析与可视化方面自主研发的VARxMD系统,一方面扩展了物理时空性质演化的分析功能,使得VARxMD可分析ReaxFF MD模拟体系中同时包含的物理和化学变化的演化过程;在此基础上进一步扩展了VARxMD聚焦于模拟体系3D局部区域内化学反应分析追踪和物理性质的分析能力。本文概要介绍VARxMD所扩展ReaxFF MD局部区域反应追踪和物理性质动态分析的建立方法,并通过交互绘制矩形选择局部区域进行反应追踪来揭示煤热解体系中煤颗粒之间孔道的作用、以及通过采用球体局部区域计算区域内径向分布函数变化进而分析对含能材料热解过程中所形成的富碳团簇结构进行表征,展示本工作扩展方法的应用及其可能的应用前景。
2 ReaxFF MD模拟体系局部区域反应追踪及物理性质动态分析方法
从聚焦局部反应区域如表面、反应热点等角度出发,在VARxMD自动分析模拟体系全局化学反应行为的基础上,我们为VARxMD程序系统扩展了聚焦所模拟体系中局部反应行为、并追踪其物理性质和化学反应随时间的动态演化规律的能力。图1是本工作扩展的聚焦ReaxFF MD模拟体系局部区域反应分析、物理性质与VARxMD全局区域的关系图。
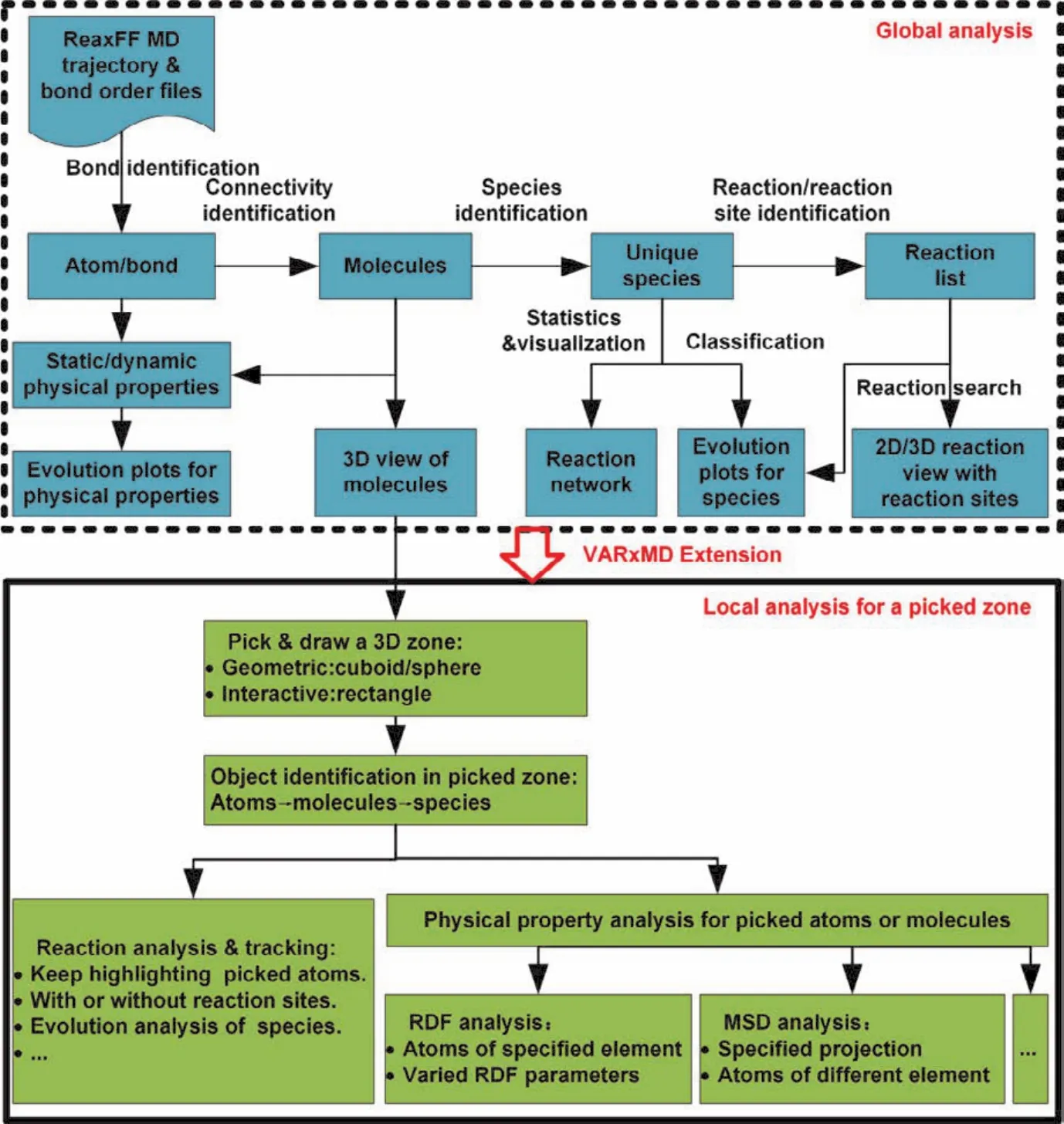
图1 VARxMD系统功能扩展:聚焦ReaxFF MD模拟体系局部区域反应追踪及物理性质动态分析Fig. 1 Schematic diagram for the extension of VARxMD: tracking local reactions and dynamic evolution of physical properties in a 3D area picked up in a simulation cell of reactive molecular dynamics.
如图1所示,针对ReaxFF MD模拟体系,VARxMD扩展的局部区域分析模块建立在已有的全局化学反应分析和物理性质动态分析模块之上。基于VARxMD全局反应分析和全局的径向分布函数RDF (Radial Distribution Function)、均方位移MSD (Mean Square Displacement)分析,局部区域分析包括指定局部区域内唯一物种间化学反应的分析追踪、以及针对特定原子和分子的物理性质分析。
要聚焦局部区域的分析,首先需要对模拟体系中特定的空间进行交互式指定,即进行局部区域空间的选取;接着程序系统自动识别区域内的原子或分子片段;再进行局部区域内以基于物种唯一性的分子片段之间化学反应的识别和分析追踪、及针对该区域内原子和分子的物理性质分析。局部区域的分析与VARxMD已有的全局分析功能平台密切相关,且其实现策略直接关系到应用于大规模模拟体系的可行性。局部区域空间的选取本质是ReaxFF MD模拟体系的模拟盒子内一个3D子空间的指定与选取,因此局部区域内的化学反应追踪与物理时空性质分析建立在VARxMD已有的模拟体系全局3D视图之上。3D局部区域分析的实现主要包括3个部分:(1)3D局部区域空间的选择与绘制;(2)局部区域内原子、分子、物种和反应的识别;(3)局部区域化学反应的追踪与物理性质动态分析。
2.1 ReaxFF MD模拟体系3D局部区域选择绘制与分子识别
要分析模拟体系中局部空间内复杂物理变化与化学反应随时间的演化规律,首先要进行3D局部区域的选择和绘制。在局部区域化学反应追踪与物理性质动态分析中,针对不同ReaxFF MD模拟可能的应用,如表界面反应、局部的反应热点、局部的团簇生成反应等,设计和实现了两类选择和绘制3D局部区域的模式。一类是用户在模拟盒子中指定3D局部空间的参考点并交互输入其几何参数后自动绘制几何体的几何模式,几何体的形状目前包括长方体和球体;由于选择与绘制区域方法是基于面向对象的C++编程模式和模块化编程实现的,若要扩充新的局部区域选择几何体例如圆柱体,可方便地加以实现。另一类交互模式是用户在3D视图上利用鼠标绘制任意大小的矩形框,经过光线投射算法识别矩形框后面的实物,再根据实物位置数据绘制长方体。由于VARxMD的3D视图可视化基于VTK11框架开发,从3D视图的指定局部区域内识别区域大小并获取数据的过程可映射为一种消息响应机制。第一类区域选择方法采用VTK可视化管线机制自动渲染出几何体空间,其伪代码如图2所示。为使VTK窗口交互得到区域数据的渲染功能满足“高内聚低耦合”的设计要求,第二类局部区域选择方法采用观察者设计模式(Observer Design Pattern)与命令设计模式(Command Design Pattern)相结合的策略加以实现,该模式可高效地交互拾取3D视图中的选中区域对象。

图2 VARxMD从模拟盒子选取长方体/球体局部区域并识别其中的分子的算法伪代码Fig. 2 Pseudo code of VARxMD for picking up a 3D area of cuboid/sphere in a simulation cell and for the identification of molecules therein.
VARxMD扩展的第二类局部区域选择方法与第一类相比,更加灵活。用户可在3D可视化界面上利用鼠标绘制任意大小的矩形、由程序系统通过光线透射算法识别出射线与矩形框相交的分子。由于用户在电脑屏幕上选择的是2D平面,该平面背后的空间实际上并不可见。为此,第二类局部区域选择的难点在于如何通过光线透射算法选择出2D平面后的矩形区域、及识别该区域内所有的分子,并对其进行高亮显示。VTK框架中的观察者设计模式可让观察者相关的目标响应同一个动作指令;命令设计模式可封装命令,并让选中的所有目标作为命令接受方,执行VARxMD用户通过屏幕鼠标触发的具体命令及操作。结合VTK框架中的观察者和命令设计模式,VARxMD可高效选中用户指定的局部区域和该区域内包含的所有分子。其实现策略如图3所示。
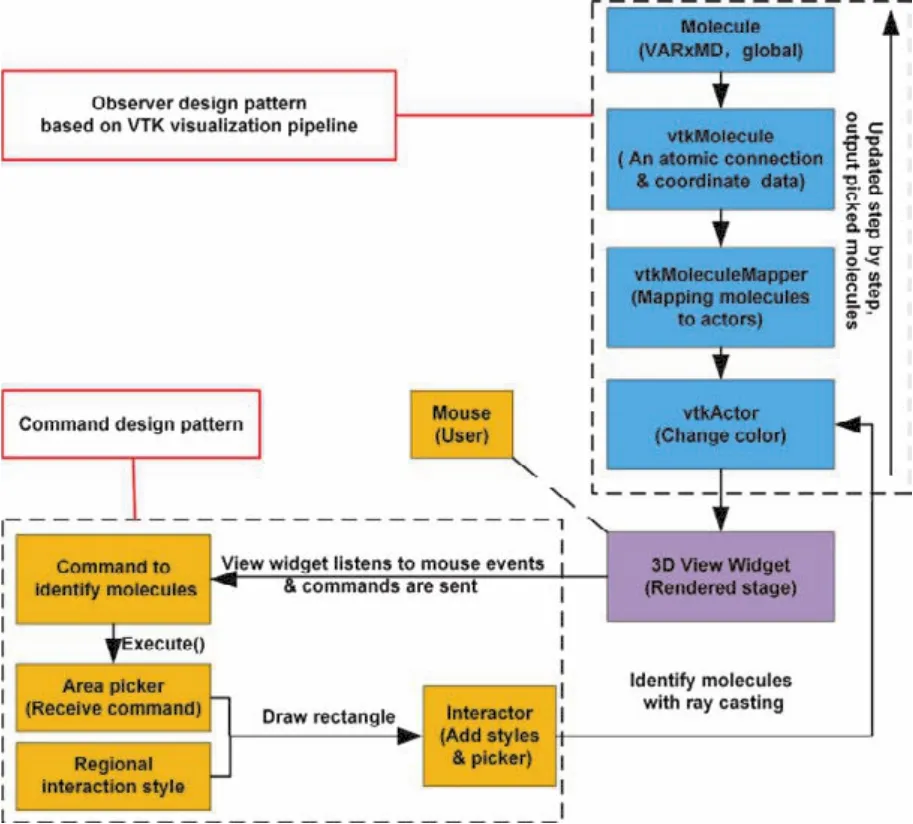
图3 VARxMD基于观察者与命令模式结合实现鼠标在屏幕交互绘制矩形选择3D局部区域实现策略Fig. 3 Implementation strategy for 3D zone pick up interactively in a ReaxFF MD simulation cell using a combination of Observer pattern and Command pattern.
如图3所示,在VTK 3D可视化编程环境19下,VARxMD通过VTK可视化客户端模块监听和接收真实用户的屏幕矩形绘制请求。基于观察者模式,将区域选择与平面矩形绘制操作建立绑定关系,选择指定区域,获得区域几何体的相关数据;接着利用命令设计模式,接受从该区域背后识别出的分子数据,并对选择出的分子改变颜色;然后沿着可视化管线的逆方向逐级更新到VTK分子数据源,将VTK分子数据源索引、继承并搜索由VARxMD全局反应分析获得的全局分子列表,从而实现了指定局部区域内的分子识别。局部区域分子识别的实现是进行指定局部区域反应分析和追踪的基础和关键。
2.2 3D局部区域唯一物种间的反应分析与追踪
当从ReaxFF MD模拟盒子中交互地绘制和选择了指定的3D局部区域、并对该区域的分子进行识别之后,就可以对该局部区域内当前时刻t的物种(即每一个区分了同分异构体的3D分子结构)与某个时刻t + ∆t(当∆t> 0为正向追踪;当∆t< 0为逆向回溯)之间的化学反应进行分析和追踪。图4给出了VARxMD的3D局部区域反应分析与追踪的算法伪代码,其中输入是从指定3D局部区域中识别出的分子和VARxMD已分析获得的模拟体系完整反应列表,输出为当前时刻t至t + ∆t之间指定局部区域所有分子涉及的化学反应。如图4所示,先遍历∆t中每两个相邻时间步的时段,在每个时段遍历当前时刻局部区域所识别出的分子集合,判定分子处于所选择局部区域的判据是该分子的质心位于该局部区域内,由此得到每个分子及其所属原子集合;再遍历原子集合,得到每个原子在相邻时刻的所属分子,并对所属分子去重;最后以当前分子和下一个(或前一个)时刻的分子联合为索引,在全局反应列表中查找化学反应,如此循环,即可获得局部区域的化学反应列表。

图4 VARxMD对ReaxFF MD模拟盒子中局部区域分子的化学反应分析与追踪算法伪代码Fig. 4 VARxMD pseudo code of reaction analysis and tracking for a picked 3D area in ReaxFF MD simulation cell.
2.3 3D局部区域物理性质RDF动态分析
VARxMD局部区域物理性质动态分析是指从物理性质的角度分析ReaxFF MD模拟体系中指定局部区域的瞬时结构特征及其演化行为。通过引入经典分子动力学模拟静态结构的径向分布函数和均方位移函数等时空性质的计算方法,定量描述所模拟的反应分子体系空间密度随时间的演化和自扩散行为等。其中,本工作以RDF径向分布函数为例(局部区域原子MSD的算法请见Supporting Information),设计并实现了区分原子和分子、并结合多条件同时分析的方法。在指定局部区域的前提下,原子的RDF计算包括不区分元素的所有原子类型、指定元素类型的原子对;并可设置不同时刻以获得RDF随时间的动态演化规律。为了方便比较不同元素的原子在特定时刻的结构特征,VARxMD特别设计了同时计算特定时刻、指定不同元素类型的原子RDF和可视化比较的功能。多曲线的RDF绘制变量可以是时间、球壳厚度、某元素原子,能清晰揭示该指定区域内物理结构在时空尺度的演化。
图5给出了RDF分析设置界面示例,用于分析某含能材料热分解体系在1250 ps、指定的某个局部区域(区域数据保存为1250 ps. RegAtoms文件)的RDF。此设置将同时分析C-C、C-H、C-O、C-N四种原子对的RDF,球壳间隔为0.02 nm。区域不同元素原子对之间RDF计算可表征该体系指定的局部区域当前时刻的结构特征,分析结果可见3.2节。

图5 VARxMD对ReaxFF MD模拟的某含材料热分解体系局部区域在1250 ps分析不同原子对之间RDF计算及多曲线可视化比较的设置界面示例Fig. 5 Screenshot of VARxMD for RDF calculation of varied atom pairs at moment of 1250 ps in a picked 3D area in thermal decomposition of an energetic material system simulated using ReaxFF MD.
3 结果与讨论
本文在针对已有的VARxMD全局化学反应分析与可视化系统的基础上,基于VARxMD中的3D视图和可视化编程环境VTK,扩展了聚焦ReaxFF MD模拟体系中指定局部区域的反应追踪及物理性质动态分析功能。此扩展为ReaxFF MD模拟应用于复杂反应过程、揭示重点区域的化学细节和探测其物理性质提供了可能。下面两节概述该新功能应用于研究煤热解过程中煤颗粒间孔道的反应分析和含能材料热分解过程中所形成纳米碳团簇的物理性质。
3.1 VARxMD的3D局部区域反应追踪应用于煤热解过程煤颗粒孔道的相互作用
煤热解是煤的热加工利用如气化、燃烧的基础过程,了解煤热解机理可为煤清洁利用提供理论支持。由于煤热解是自由基驱动的高温快速反应过程,已有的实验手段难以原位捕捉煤热解过程中的中间体和反应细节。基于对大规模煤分子模型的ReaxFF MD模拟可揭示煤热解过程反应、中间体及热解产物的演化规律,是一种有潜力深入认识煤热解本征反应的方法20–22。
一般认为,煤热解过程起始于煤结构中最弱的桥键,桥键断裂之后,某些较小的挥发分中间体会迁移至游离空间发生二次裂解或者被小分子自由基(HO、CH3等)稳定下来,促进煤热解过程23。因此,两个煤颗粒之间的孔道(游离空间)发生的化学反应对认识煤热解过程至关重要。由于反应分析能力的限制,已有的煤热解反应模拟主要针对单个煤颗粒内部的模拟。3D局部区域反应分析与追踪为研究两个煤颗粒孔道之间的反应细节提供了可能。图6a是利用VARxMD新功能分析的一个包含了两个煤颗粒的大规模煤模型在高温的热解反应的示例。该煤模型含99544个原子,分子式为C46920H43320O9088N72S144,其中两煤颗粒间的孔道间距约为1 nm。利用ReaxFF MD,采用慢速升温速率2 K·ps−1,将该煤模型从500 K加热至2500 K。本次示例以10 ps为时间间隔,分析了从5–205 ps(510–910 K)、共20帧模拟轨迹的初始煤热解过程,共得到4782个化学反应。

图6 (a)VARxMD的屏幕交互绘制矩形选择3D局部区域应用于ReaxFF MD模拟的煤热解体系煤颗粒孔道间化学反应追踪分析的屏幕截图示例以及(b)5 ps和205 ps 3D视图的对比Fig. 6 (a)VARxMD screenshot of the 3D picked zone and the reactions tracking therein through interactive drawing of a rectangle on screen, focused on channel reactions and (b)comparison of 3D views at 5 ps and 205 ps of coal particle thermal systems simulated using ReaxFF MD.
如图6所示,利用VARxMD新扩展的屏幕交互绘制矩形模式(第二类选择模式)选择两个煤颗粒之间的孔道空间,即图中3D窗口红色边框的长方体局部区域。该长方体沿X轴方向的长度为2.612 nm,占模拟盒子X轴向长度20 nm的13.06%。长方体内蓝色高亮部分为选中的3D局部区域内所识别出的原子与键;右侧是以树图结构展示的局部区域原子所参与的化学反应列表,每个反应的细节可在反应列表树图下方以2D化学结构式清晰展示,也可通过Region Species Generator检索识别出该局部区域内所有物种的分子集合,并基于元素、分子式、基团等进行反应物或生成物的统计和归类。图6b给出了指定局部区域在5 ps和205 ps的3D视图对比,可清晰看出随着煤颗粒间的反应发生,局部区域逐渐被热解反应生成的产物和中间体分子占据,煤颗粒间的较大孔道因剧烈反应发生变小,微孔道增多。在5–205 ps可分析得到局部区域的化学反应为932个。
表1给出了在5–205 ps的煤热解初始过程中指定局部区域发生的化学反应数量与全局反应数量的对比。如表1所示,虽然所选取的孔道局部区域体积只占整个煤模型体系体积的13.06%,该区域发生的化学反应在裂解起始阶段占了全局体系的20%左右,说明该区域的确是煤热解的活跃区域,生成的中间体和自由基会促进整个煤分子的热解,增加煤焦油的收率。通过本工作实现的局部区域反应细节追踪的独特功能,可获得在该段模拟时间获得的特征化学反应及其2D结构。表2举例列出了部分反应的化学方程式,基于2D结构的化学反应表示在Supporting Information中提供,这些反应物种的2D结构信息可帮助深入认识该区域的相互作用和所发生的化学变化。如表2所示,该区域发生了大部分煤热解的特征反应,包括最薄弱的-O-CH2-桥键断裂(表2中的反应2、5、6、7、9、12、16、17),HO、CH3等游离小分子自由基的生成(表2中的反应6、8、14),煤活化的结构转化和可逆震荡反应(表2中的反应3、4),以及由于HO、CHO2脱落和H转移反应促进的H2O和CO2生成(表2中的反应10、15、16、17)。此外,由于孔道空间较大,有利于小分子自由基的移动,该局部区域也发生了单一煤颗粒分子模拟时没有发现的初始裂解过程中H2的生成(表2中的反应1)和轻质焦油的生成(表2中的反应11)。这些局部区域的反应细节有利于帮助认识孔道对煤热解的作用,是其它分析软件或扩展之前的VARxMD功能无法提供的。
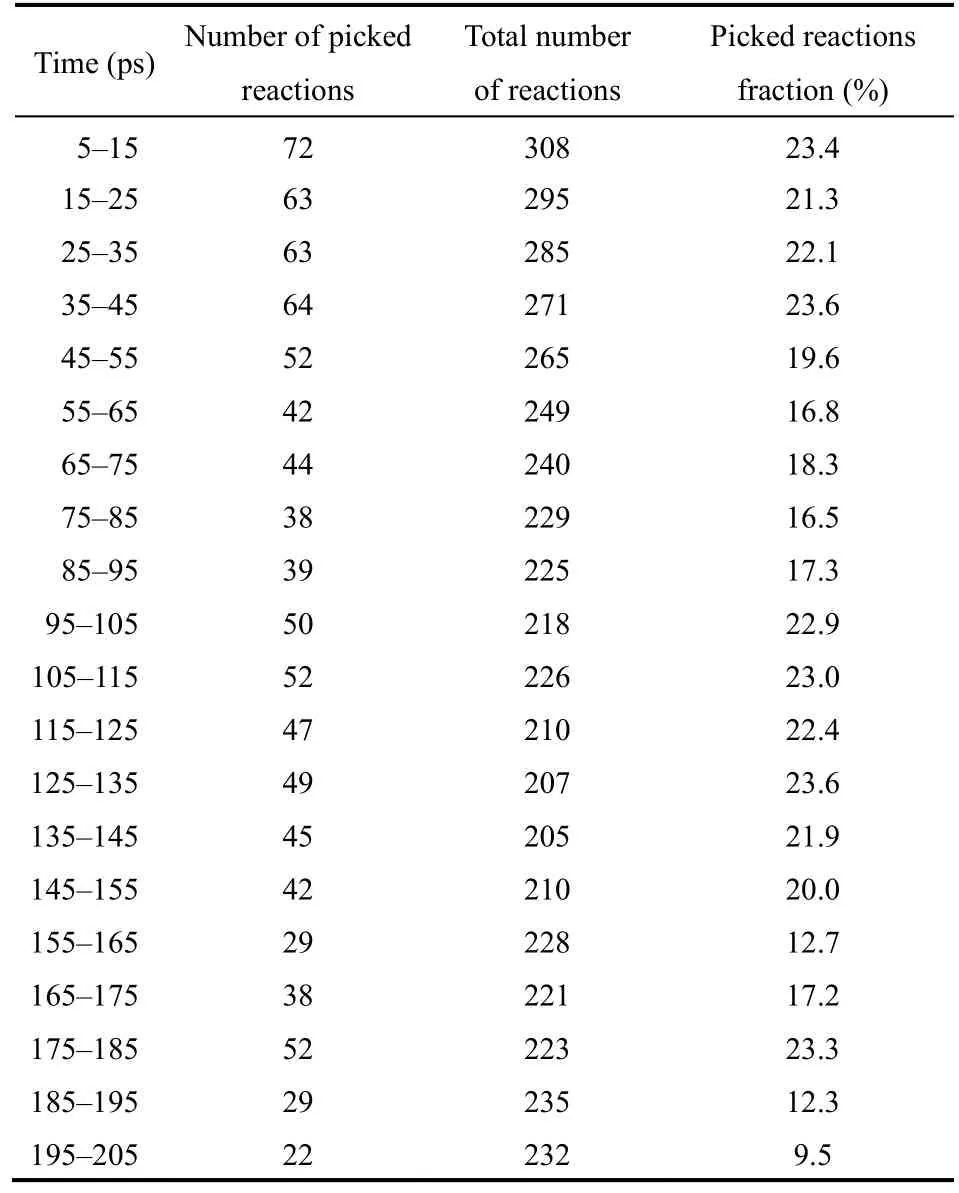
表1 图6煤热解模拟体系内煤颗粒孔道局部区域化学反应与全局模拟体系反应在5–205 ps间的数量对比Table 1 Number comparison of the local chemical reactions in the picked zone with the total reactions in thecoal pyrolysis simulation system of Fig. 6 during the period of 5–205 ps.

表2 5–205 ps模拟时间内煤颗粒体系特征化学反应列表Table 2 Examples of characteristic reaction of coal particle system during 5–205 ps.
3.2 VARxMD的3D局部区域物理性质动态分析应用于含能材料热解过程的团簇生成
近年来,基于ReaxFF力场的反应分子动力学方法在认识含能材料复杂反应行为的研究非常活跃,从微观尺度揭示了多种含能材料在热载荷24、压缩25、冲击26,27以及剪切28等不同外部刺激下的化学响应机制,而且为了解炸药在高温高压极端爆轰条件下复杂化学过程提供了可能。团簇大分子是富碳含能材料TNT爆轰过程中的重要产物,与炸药的爆轰过程和做功能力密切相关,是含能材料爆轰领域的研究热点29。以利用ReaxFF MD模拟研究大规模TNT晶体模型(经验化学式为C8960H6400O7680N3840)在高温3000 K的热分解-膨胀-冷却过程为例,借助VARxMD最新扩展的局部区域物理性质动态分析功能,可帮助认识TNT高温热分解过程中富碳团簇的生成机理。该模拟体系含1280个TNT分子,共26880个原子,模拟的总时长为1350 ps,时间步长0.1 fs,轨迹输出间隔为100 fs。利用VARxMD以较大的时间间隔25 ps分析完整的模拟轨迹,可获得5150个反应。
在TNT高温热分解模拟的末期可观察到许多大小不同的原子团簇形成(如图7左侧所示,模拟盒子在1250 ps时刻的3D截图)。为细致观察所生成原子团簇,在VARxMD的3D可视化窗口中交互选择了模拟盒子中一处包含层状结构的球形区域(半径为2 nm,如图7右侧所示)作为重点分析的对象。该局部区域内包含几层大富碳分子碎片及一些CO2,N2,H2O,H2等小分子气体产物。为进一步分析该层状团簇的结构特征,计算1250 ps时刻下该局部球形区域内不同原子的径向分布函数。首先分析了选中局部球形区域内C-C原子对的RDF曲线(间隔参数为0.002 nm)以了解原子团簇碳环骨架的结构特征,如图8a所示。与5层石墨烯结构的C-C原子对RDF进行对比,可以看到该层状分子团簇的C-C原子对的RDF主要峰值位置和石墨烯吻合较好,这表明该层状分子团簇具有类石墨烯结构。观察该层状原子团簇各片层分子的结构(如图8b所示),可以看到每一分子碎片的主体骨架均具有稠环结构,以六元碳环为主、夹杂少量五元和七元碳环结构,每一分子片层的周围还带有一些侧链基团。这些结构特点说明该层状分子团簇尚未形成完美的石墨烯六元稠环结构,仍需更长时间的演化。
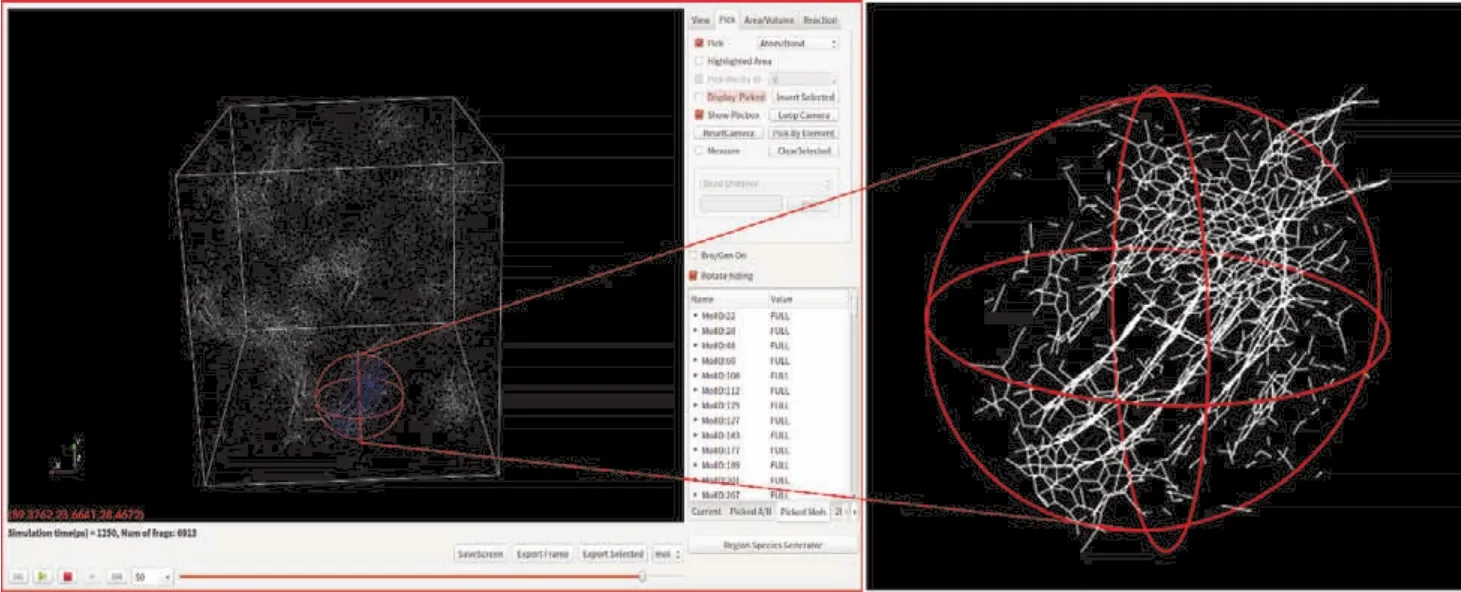
图7 VARxMD的绘制球形选择3D局部区域功能应用于ReaxFF MD模拟TNT热解体系在1250 ps的界面截图Fig. 7 VARxMD interface screenshot of drawing a sphere and selecting 3D local area function applied to ReaxFF MD simulation TNT pyrolysis system at 1250 ps.

图8 (a)TNT晶体模型热解体系的球形区域层状分子团C-C的RDF与5层石墨烯C-C的标准RDF对比及(b)层状团簇中主要大分子结构Fig. 8 (a)Comparison of the RDF of the spherical region layered molecular clusters C-C of the pyrolysis system of the TNT crystal model with the standard RDF of the 5-layer graphene standard C-C and(b)major macromolecular structures in layered clusters.
选中的层状结构的大分子团簇中还含有一些N、O杂原子,这与已报道的富碳含能材料爆轰得到碳纳米材料中常含有N、O杂原子是吻合的30。因此利用新扩展的局部区域多条件RDF分析功能,计算了区域内其他原子对C-O、C-N和C-H之间的RDF(间隔参数为0.02 nm),如图9所示。由不同原子的径向分布函数曲线的最大峰值可以看到,C-O、C-N和C-H键的键长分别在0.13 nm、0.13 nm和0.11 nm左右。与标准原子间共价键键长比较,可确定团簇中C和O原子之间多为单键,而C和N原子间主要为双键。
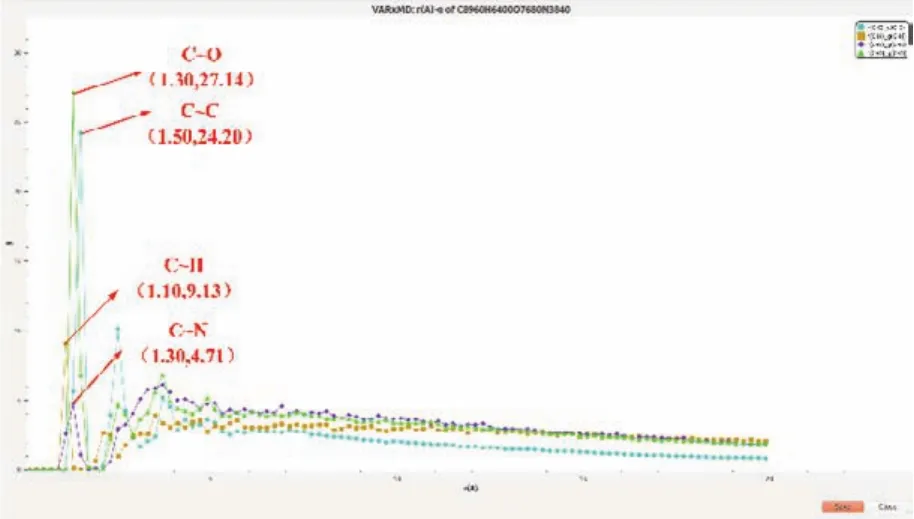
图9 VARxMD分析ReaxFF MD模拟的TNT晶体模型热解体系的球形局部区域在1250 ps,0.02 nm下的不同原子对之间RDF计算结果的显示界面截图Fig. 9 VARxMD analysis screenshot of the display interface of the RDF calculation results between different atomic pairs at 1250 ps, 0.02 nm in the spherical partial region of the TNT crystal model pyrolysis system simulated by ReaxFF MD.
4 结论
本工作基于自主研发的、先进的反应分子动力学模拟结果反应分析与可视化工具VARxMD程序系统,以VARxMD的3D可视化视图作为切入点,为VARxMD进一步扩展了对指定的3D局部区域内的进行反应追踪与瞬态物理性质分析能力。从ReaxFF MD模拟体系全局分析到3D局部区域分析能力的扩展使得研究者聚焦分析模拟体系内、特定的局部反应热点成为可能,已应用于煤热解反应模拟中煤颗粒间的反应追踪以及含能材料热分解过程中富碳团簇形成中的瞬态结构表征,也有望应用于催化反应体系表界面反应分析、含能材料爆轰过程中反应热点分析等复杂反应过程的研究。
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
猜你喜欢
杂志排行

物理化学学报的其它文章
- Photocrosslinking-Immobilized Polymer Vesicles for Lowering Temperature Triggered Drug Release
- Poly(ε-caprolactone)-Polypeptide Copolymer Micelles Enhance the Antibacterial Activities of Antibiotics
- 稀土-天然皮革可穿戴X射线防护材料的合成及性能
- 石墨烯玻璃透明薄膜加热特性
- Single-Molecule Field-Effect Transistors with Graphene Electrodes and Covalent Pyrazine Linkers
- 红色荧光碳量子点用于肿瘤微酸环境诊断