表面限域掺杂提升高比能正极材料稳定性
2021-11-22张思东刘园祁慕尧曹安民
张思东,刘园,祁慕尧,曹安民,*
1中国科学院化学研究所,分子纳米结构与纳米技术院重点实验室,北京分子科学国家中心,北京 100190
2中国科学院大学,北京 100049
3郑州大学基础医学院纳米酶医学中心,郑州 450001
1 引言
随着化石燃料的逐渐消耗和环境的持续恶化,发展环境友好且可持续的能量储存技术成为可持续发展的必然需求1。与铅蓄电池、镍-氢(Ni-H)电池以及超级电容器等能量存储技术相比,可充电的锂离子电池在能量密度上具有显著优势2,3。因此,自1991年锂离子电池商业化之后,其在便携式电子设备等方面得到了广泛应用4,而长续航里程电动汽车、智能电网等领域的兴起与发展则对下一代高能量密度、高循环寿命的锂离子电池提出了新的要求5,6。与之相适应,具有更高能量密度的新型电极材料的开发成为提升锂离子电池电化学性能的重点与难点7,8。
在锂离子电池中,正极材料是决定电池能量密度的关键因素,锂离子电池的发展依赖于具有高能量密度、高循环稳定性、高安全性且价格低廉的正极材料的设计与开发9。自LiCoO2(LCO)作为第一代正极材料得到广泛应用后,一系列具有不同晶格结构的正极材料,如层状、尖晶石和橄榄石型等相继投入应用,均在可逆脱嵌锂离子、提供高能量密度电池上表现出广阔的应用前景10–13。高比能正极材料通常表现为高工作电压或高比容量(能量密度=平均电压×比容量),因此,从电极材料的结构设计出发,结合材料自身晶型、组分、形貌等多方面的调控,获得具有高可逆容量或高工作电压的正极材料成为高比能电池开发的重要研究内容和发展方向。
以高镍三元、富锂锰基、高电压尖晶石等为代表的氧化物正极材料是下一代高比能正极材料研究的热点14。其中,具有层状晶格的高镍三元材料其大规模商业化应用已逐渐展开,该材料化学式为LiNixMnyCozO2(NMC,x+y+z= 1,如NMC811材料),其高镍含量有可能提供高于200 mAh·g-1的可逆比容量,在长续航动力电池电极材料上具有显著优势。NMC正极材料中的Ni、Co、Mn三种过渡金属元素可以以适当的比例相互掺杂,形成LiMO2型层状材料(图1a)。不同的过渡金属发挥不同的功能15:镍基于Ni2+/Ni3+、Ni3+/Ni4+的氧化还原电对为活性材料提供高的可逆容量;钴也作为电化学活性元素存在,同时它还可以抑制阳离子重排,从而稳定材料的晶格结构;Mn4+已经处于高价,虽然它不提供容量,但它不仅能够起到稳定晶格结构、提高安全性的作用,同时还有利于降低材料成本。NMC中三种金属的比例可调,所对应的正极材料自身物理化学特性也将存在显著差异。此外,富锂锰基层状正极材料xLi2MnO3·(1-x)LiMO2(M为Ni、Co、Mn、Fe等)具有较高的比容量(超过250 mAh·g-1),而尖晶石材料LiNi0.5Mn1.5O4能提供4.7 V的高工作电压,两者也成为目前高比能正极材料研究的热点。

图1 (a)NMC组成图;(b)层状三元材料NMC的放电容量和热稳定性之间的关系16Fig. 1 (a)NMC composition diagram; (b)Correlation between discharge capacity and thermal stability of layered structure ternary NMC 16.
近年来,随着正极材料比能量的提升,其自身的稳定性、安全性问题也日益显著。例如,对于NMC而言,随着材料中镍含量的增加,其比容量会相应增加,但材料的热稳定性将出现逐渐降低的趋势(图1b)16。与此同时,随着镍含量的增加,电池的副反应风险、循环稳定性将存在更大的挑战17。这种不稳定状态不仅取决于NMC自身的物化特性,还与循环过程中正极电解质界面(cathode electrolyte interphase,缩写为CEI)的稳定性密切相关。在脱锂状态下,NMC中的过渡金属将被氧化而处于高价态(如镍将主要以Ni4+存在),此时的高价镍离子具有很强的氧化性,很容易与有机电解液发生氧化还原反应,导致出现电解液分解、产气等一系列问题16,18;值的注意的是,随着电化学反应的持续发生的进行,相关问题将逐渐加剧,长循环过程中电极材料的稳定性、安全性问题将更为显著19。事实上,基于高镍三元材料的动力电池事故时有发生,已经成为高镍材料大规模应用的严峻挑战。从电极材料的结构设计出发,开发可靠的结构稳定策略以提升高比能电极材料稳定性,已经成为正极材料研究的迫切需要与共识。
鉴于CEI对电池稳定性、安全性的重要意义,从正极材料自身表面结构调控出发,提升CEI的稳定性成为获得高比能、高稳定锂离子电池的重要手段。本文将围绕不同正极材料的失稳机制展开讨论,对提升高比能正极材料稳定性的途径进行综述和总结;在此基础上,着重介绍表面限域掺杂这一新近发展的稳定机制,以期获得对正极材料表面稳定原理的新认识和新途径。
2 高比能正极的失稳途径
2.1 环境因素影响
对于高镍三元NMC和富锂材料而言,其储存过程中往往因为环境因素的影响而造成结构坍塌和性能衰减,这一失稳途径在高镍三元正极上更为显著。高镍三元中Ni主要以Ni3+存在,其自身化学性能不稳定,极易自发的被还原成Ni2+而造成结构破坏;同时,材料表面的锂也容易与空气中的CO2和H2O发生副反应,生成无定形的Li2CO3和LiOH,从而导致不可逆的锂损失20。例如,在近期的研究中,Wang等人21利用高分辨透射电镜(HRTEM)等手段确认水蒸气易触发高镍三元材料的脱锂过程,而CO2的存在则会加剧这一过程。如图2中HRTEM的横截面所示,当NMC811分别暴露在O2、N2、CO2和水蒸气中30 min后,沿[010]区域轴看,O2、N2并未对NMC811表面起到腐蚀作用(图2a,b);而将NMC811暴露于水蒸气中30 min后(图2d),NMC811表面有几个原子层厚度的新相形成,表明水蒸气容易激活材料的界面反应;此外,单独的CO2气氛中暴露并未在NMC811表面形成Li2CO3(图2c),表明CO2自身不会对正极材料表面产生腐蚀作用,只有在与水蒸气共存时才会将LiOH钝化层转化为Li2CO3,并造成正极材料在空气中的持续破坏。

图2 (a–d)上图为NMC 811表面在气压为5 × 10-2 Torr和室温下分别暴露于N2、O2、CO2和水蒸气30 min后的HRTEM图像;下图显示在各相应气体暴露下层状结构中Li+的演化示意图21Fig. 2 (a–d)The upper panels are the HRTEM images of NMC 811 surface after exposure to N2, O2, CO2, and water vapor at the gas pressure of 5 × 10-2 Torr and at the room temperature for 30 min,respectively. The lower panels show the schematic drawing of Li ions evolution in the layered structure under the exposure of each corresponding gas 21.
对于NMC正极而言,在环境中暴露造成的正极结构破坏会对其电化学性能发挥造成显著影响:首先,无定形组分的存在会阻碍离子和电子的运输,导致电极材料的容量损失和极化加剧;其次,强碱性物质的存在(pH接近11)对于电极制备过程中的打浆工艺不利,作为粘结剂的PVDF在强碱性条件下会发生脱氟化过程,从而失去粘结剂的粘结功能22;此外,Li2CO3等杂质会在高电压下分解产生气体如CO2、CO等,这会给电池的实际应用带来很大的安全隐患23。相对于NMC正极尤其是高镍三元正极材料,富锂层状材料以及高电压尖晶石镍锰酸锂正极材料在环境中的结构稳定性较好,因此在材料存储以及电池组装过程中对环境湿度的要求也相对较低。
2.2 循环过程中的结构衰减
正极材料在电化学过程中会发生持续的结构变化,导致其自身结构的破坏及电化学性能的衰减。而随着高比能正极循环次数的增加,其结构不稳定性及安全风险也逐渐加大。
对于高比能正极材料而言,层状富镍材料中,过渡金属位于八面体的3b位置,而Li+位于八面体的3a位置24,25。根据晶体场理论,镍离子的+2价结构稳定,而由于Ni2+(0.069 nm)和Li+(0.076 nm)的半径相接近,在这种情况下,材料中的Ni2+容易迁移到Li+的3a位置,造成阳离子混排的现象26,27。在电池充电过程中,由于占据锂层的镍具有电化学活性,随着镍价态的升高,离子半径变小,这将会阻碍锂离子的扩散通道(如图3a所示)28,最终造成富镍正极层状结构逐渐演化为电化学惰性的岩盐相,导致富镍材料的电化学性能快速衰减。而如果向其中引入与Ni2+半径相似的惰性金属离子如Mg2+,Mn4+等,锂离子的扩散路径则不会被阻碍29,30。如Cho等人30发现,表面掺杂Mn的LiNi0.62Co0.14Mn0.24O2材料因为支柱层的存在而变得更加稳定(图3b)。

图3 在LixNi1+ZO2体系,(a)第一个电化学循环过程中Ni2+离子的氧化引起空间局部塌陷,这使锂扩散和重新嵌入变得困难28;(b)LiNi0.7Co0.15Mn0.15O2, LiNi0.6Co0.2Mn0.2O2和LiNi0.62Co0.14Mn0.24O2的电化学循环性能30Fig. 3 In the LixNi1+ZO2 system, (a)during the first electrochemical cycle, the oxidation of Ni2+ causes a local collapse of the space, which makes it difficult for the diffusion and re-intercalation of lithium ions 28;(b)Electrochemical cycling performances of LiNi0.7Co0.15Mn0.15O2, LiNi0.6Co0.2Mn0.2O2 andLiNi0.62Co0.14Mn0.24O2 30.
除Li+/Ni2+混排外,电解液分解造成的正极材料结构中金属离子的溶出也是导致电池电化学性能衰减的主要原因之一。电解液中往往存在微量的水,水的存在容易使电解液发生如下分解反应31:LiPF6⇌ LiF + PF5;PF5+ H2O → POF3+ 2HF。在高电位下,Ni4+的存在会进一步加剧电解液的分解32。电解液分解产生的强酸性HF极易腐蚀电极材料,容易造成材料中的过渡金属离子如Mn2+的溶出。锰离子的溶出会产生两个负面影响:首先溶解于电解液的Mn离子会重新在正极材料表面沉积,形成电子和锂离子的阻碍层。而溶出的Mn离子也会通过电解液迁移到负极,在负极表面的过渡金属会催化固体电解质界面(solid electrolyte interphase,缩写为SEI)的分解,从而阻碍稳定SEI的形成(图4a)33。电解液氧化分解产生的酸性物质也会加速SEI中烷基碳酸锂的分解(图4b),使SEI层出现裂纹以及非活性层的形成,在充放电循环过程中SEI层厚度不断增加,界面阻抗不断增加,最终导致电化学性能衰减34。
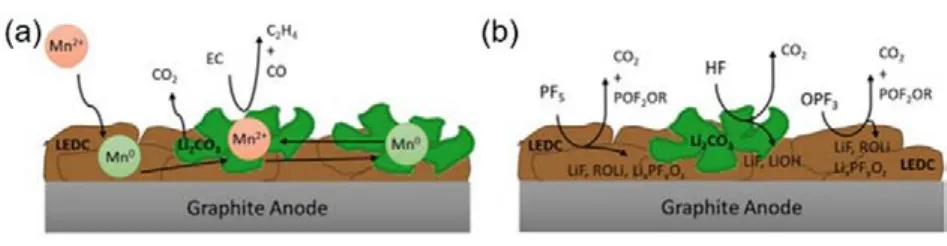
图4 (a)锰催化负极SEI分解的示意图33;(b)酸性物质诱导负极SEI分解的示意图34Fig. 4 A schematic representation of the (a)manganese catalyzed degradation of the anode SEI 33; (b)Acidic species induced decomposition of anode SEI components 34.
电池充放电时,锂离子会不断地在正极材料中嵌入和脱出,在锂离子脱嵌过程中会引起材料在a、b、c三个方向上的各向异性膨胀/收缩35。随着循环次数的增加,颗粒之间容易形成晶间裂纹,颗粒内部容易形成晶内裂纹。由应力产生的晶间裂纹容易使得正极材料颗粒之间逐渐失去电接触,而且会产生更多的新的表面暴露在电解液中,进一步恶化电池电化学性能36。Novák等人37报道表明晶间裂纹的产生情况与截止电压和循环次数有关,截止电压越高,循环次数越多,晶间裂纹就越多。除了晶间裂纹,Wang等人36在长循环后的NMC111材料中发现大量的晶内裂纹。作者通过系统地研究并借助扫描透射电镜(STEM)技术很清晰地观察到裂纹的成核、生长、驱动扩散的过程,发现晶内裂纹是由电化学驱动和扩散控制的,并提出基于晶粒内部位错的裂纹孵育机制。此外,从平面内方向来看,断裂行为的起源被认为是由于过渡金属和氧键之间的伸长引起的平面外延生长。Lin等人38根据文献报道将裂纹的产生所带来的影响主要归为如下四类(图5):(1)活性颗粒之间差的电接触;(2)裂纹容易产生失去电接触的碎片,造成活性材料的损失;(3)晶间裂纹的扩展会暴露新的界面,加速正极-电解液界面的副反应;(4)过渡金属的溶出。对于这四个主要负面影响,它们之间并不相互独立,锂离子电池衰减的电化学性能主要是这四个负面因素的协同效应影响的结果。

图5 通过晶间裂纹和晶内裂纹诱导的四种具有代表性的不利影响38Fig. 5 Schematic representation of the four negative impacts induced by intragranular and intergranular cracks 38.
对于富锂锰基正极材料而言,结构中阴离子(O2-)也参与电化学氧化还原反应,是其高容量的来源。然而这个过程常常伴随着不可逆的晶格失氧,从而导致较快的结构及容量衰减39。通过引入第四及第五周期的过渡金属(TM)离子,可以增加TM-O的杂化程度,从而稳定O2-/O2n-的氧化还原电对,减少氧气的析出。此外,在富锂材料的充电过程中,O2-被氧化的同时,TM层中的Li会迁移到相邻的Li层中,而其他位置的TM离子则会迁移填充部分Li的空位,以补偿脱锂导致的电荷分布不均40。这种阳离子重排的过程将会打破TM层的短程有序结构,而持续的TM离子迁移到Li层的过程,会促使材料从层状结构转变为尖晶石结构,这一不可逆的过程导致了电化学性能的持续衰减以及电位的下降36,41。为了使材料中的氧能够参与氧化还原反应,通常富锂锰基正极材料的充电截止电压都在4.6 V以上,较高的电压使得常规的碳酸酯类电解液在电极表面更容易发生氧化分解,从而导致电解液的不断消耗以及电池的极化不断增大。此外,不可逆的相转变以及晶格失氧的过程通常在电极材料的表面优先发生,使得表面Mn4+更容易被还原溶出,进一步加剧了材料结构及性能的衰变42,43。这就使得材料表面修饰的策略成为提升稳定性的最有效方法之一。
另一种高比能正极材料:尖晶石LiNi0.5Mn1.5O4材料,其4.7 V的工作电压为其带来了高达640 Wh·kg-1的能量密度,与富锂材料类似,在更高的工作电压下,电解液更易分解侵蚀电极材料表面,正极-电解液界面的稳定性更差,电解液自身也更容易分解44。同时,尖晶石材料中存在的Mn3+引起的Jahn-Teller效应以及自身歧化反应导致Mn2+的溶解,都使得材料的循环稳定性,尤其是高温下的循环及结构稳定性较差45。
3 表面限域掺杂稳定正极表面
鉴于高比能正极材料所面临的稳定性、安全性风险,对材料结构进行进一步调控与优化已经成为电极材料制备中的一个必要环节。对材料进行表面包覆处理,能够减缓材料的腐蚀和电极材料/电解液界面的副反应,从而延长材料的寿命和提高电池的安全性46。与此同时,对电极材料的体相晶格进行取代可以提升晶格的稳定性并调节材料的化学属性,从而有利于维持稳定的结构。因此,表面包覆和体相掺杂已经成为正极材料改性的重要研究内容,相关工艺也成为正极材料生产和应用的关键工艺。
值得注意的是,无论是表面包覆处理还是体相掺杂取代,其对材料稳定性的提升效果均有局限性。表面包覆层的构筑有助于稳定电极材料与电解液之间的界面,以此抑制界面的副反应,然而惰性包覆层自身不利于电荷的传输,包覆层的控制及高倍率性能的实现成为了电极材料表面处理的挑战47;此外,金属离子的掺杂取代有助于提升电极材料的稳定性,但某些掺杂离子的浓度必须控制在较低水平以免牺牲材料的容量,且掺杂对结构稳定的能力有限48。
从稳定表面和维持容量的需求出发,只对电极材料表面(一般小于20 nm深度)进行高浓度掺杂,一方面这种掺杂能够显著提升表面晶格结构的稳定性,同时由于掺杂不涉及体相晶格的变化,从而不影响体相容量的发挥,以此可以基于这种表面限域掺杂的方式获得兼具高稳定性、高容量的正极材料。此外,这种将正极材料表面层转化为高浓度掺杂层的方法与构筑一个惰性包覆层(如Al2O3)既有区别,又存在着一定的联系。首先,表面高浓度掺杂后能加固表面的晶格结构,提升其结构稳定性和电化学稳定性,通过调控掺杂效果,使得获得的稳定表面层仍具备一定的离子传输能力,这较之于常规的不具备电化学活性的惰性包覆层具有显著优势,能够不影响材料倍率性能的发挥;同时,表面限域掺杂的实现又往往有赖于惰性包覆层的构筑。为实现限域掺杂的效果,需要先在正极材料表面包覆上含有目标掺杂金属离子的纳米层,再结合高温固相反应,使得掺杂离子可控渗入表面的纳米深度,获得掺杂的效果。需要指出的是,在进行表面处理效果评价时,由于表面的不均一和缺乏严格确定的表面状态,不同的研究者得到的实验结果和提出的机理会有所差异。因此,构筑表面晶格结构精确可控、表面化学属性均一的电极材料,对厘清表面稳定机制、优化表面稳定能力有重要意义,这也对表面层构筑的能力提出了更高的要求。
图6简要描述了获得表面限域掺杂效果的处理路线49。通常包含两个步骤:为了获得表面均一化处理的效果,首先在正极材料颗粒表面构建一个均匀的纳米层,然后在高温条件下引发颗粒表面与包覆层之间的固相反应,这将导致外层纳米层的消失并使金属离子进入表面晶格中形成掺杂层。通过调控表面固相反应的参数,如反应物种类、温度、反应时间等,可以实现表面掺杂效果的调节。

图6 表面限域掺杂层的形成示意图49Fig. 6 Schematic diagram of the formation of a surface-limited doped layer 49.
基于表面限域掺杂,可以使得高比能正极获得具有更高稳定性的表面,同时由于表面掺杂只局限于有限的纳米深度,具有微米粒径的正极颗粒体相并未受到影响,从而正极材料自身的容量能得到充分发挥,这样能够获得兼具表面包覆、体相掺杂优势的表面稳定效果。目前,一系列不同的正极材料均进行了相关的表面掺杂尝试,在提升电极材料电化学性能上展示了显著的效果,证明这种基于表面限域掺杂的策略来稳定正极材料的表界面是行之有效的,相关的数据如表1所示50-65。

表1 不同正极材料通过不同金属的表面限域掺杂的电化学性能Table 1 The electrochemical performance of different cathode materials through the surface confinement doping of different metals.
4 表面限域掺杂稳定机制
表面限域掺杂效果的实现有赖于表面包覆物种与正极材料材料之间的高温固相反应。对于不同的正极材料而言,它们具有不同的晶格化学属性,从而发生高温固相反应时反应活性也各有不同;此外,掺杂离子的种类、浓度、固相反应的温度、时间也对最终的表面反应方式产生影响。因此,不同类型的正极材料表面掺杂效果会具有不同的表现形式。本文将结合课题组自身的工作经验,以尖晶石LiNi0.5Mn1.5O4(LNMO)材料为主要研究对象,讨论高比能正极材料在表面限域掺杂时所存在的几种结构稳定状态。
4.1 表面元素取代
富锂层状结构氧化物因高的能量密度而被认为是最有应用前景的正极材料。但是该材料在电池循环过程中面临着严重的问题,如首圈不可逆容量损失、差的倍率性能和循环性能以及持续的电压降。中国科学院物理研究所王兆翔等63确认在Li1.2Mn0.54Ni0.13Co0.13O2(LMR)材料表面中掺杂Nb等重金属离子能够显著改善富锂材料的稳定性。研究人员对表面Nb掺杂的样品(LMR-Nb)进行了仔细的原子结构表征。图7a的球差电镜照片显示LMR-Nb颗粒的体相是清晰的层状结构,而图7b中材料表面的环形暗场像显示Nb将取代表面部分的锂位,获得Nb掺杂的表面层,表面转化为具有类岩盐的结构(图7b);另外,图7c中的环形暗场像表明LMR-Nb和LMR材料两者其重量较轻的原子排布相似,Nb的掺杂并没有在LMR-Nb材料中引起显著的晶格错位;而原子分辨能量色散谱图(图7d)显示Nb在材料的表面均匀分散,这体现了Nb在表面形成一层均匀的掺杂层(图7e)。作者基于原子结构的精确表征确认了Nb在LMR材料的表面层中进行了掺杂,且掺杂存在于表面5–6个原子层范围,体现了限域掺杂的效果(图7f)。这种表面限域掺杂的样品LMR-Nb材料在电化学稳定性上显示显著提升,材料循环100圈仍具有94.5%的容量保持率,而未掺杂样品的容量保持率只有76%。通过表面掺杂离子的引入,能够显著钝化表面的副反应活性,从而获得具有高容量、高稳定性的富锂正极材料。其他金属离子(Mg2+、Ti4+、Sn4+等)在表面的元素取代也可以实现类似的效果。

图7 样品的STEM图(a)体相的高角度环形明场(HAADF)像;(b)近表面的HAADF像;(c)b图中放大的表面环形明场相;(d)HAADF像中Mn和Nb的EDS能谱图;(e)表面掺杂层;(f)表面掺杂和Nb增强的表面结构63Fig. 7 STEM image of the sample (a)High-angle annular bright field (HAADF)image of the bulk; (b)HAADF image of the near surface; (c)the ABF enlarged image of the surface area in (b); (d)EDS spectrum of Nb and Mn in the HAADF image; (e)surface doping layer; (f)surface doping and Nb-enhanced surface structure 63.
在近期的研究中,Chen等人66利用Ni在LiMn2O4正极材料表面进行掺杂以调控正极材料表面的物理化学特性。作者结合理论计算和电化学性能表征指出,表面限域掺杂的样品能够显著降低界面阻抗,有力促进材料动力学性能的提升。值得注意的是,表面掺杂的样品在低温下仍显示了高倍率性能,充分展示了表面晶格调控对正极材料离子传输、长循环稳定性等方面的关键作用。
表面掺杂离子的引入往往通过高温固相反应实现,随着掺杂离子逐渐渗入表面晶格,有可能引起正极材料自身晶格结构发生改变,进而造成较大的晶格变化与结构变形,并最终形成具有不同相结构的表面层。我们对Zn2+在高电压LNMO表面的掺杂调控展开了系统研究,表面的晶格结构对掺杂的Zn2+浓度非常敏感(如图8所示)67。随着掺杂Zn2+浓度的增加,会引起表面尖晶石结构的连续变化,并相应的对材料的电化学性能产生影响。实验中,可以先在LNMO颗粒表面构筑一层均匀的ZnO包覆层,通过精确控制ZnO纳米层的厚度来调控用于掺杂的Zn2+的量,并结合表面原子结构的表征来考察掺杂浓度控制对晶格结构的影响。我们发现,Zn2+的掺杂容易使得尖晶石相转化为层状相,随着Zn2+浓度的增加,又会进一步生成类岩盐相;控制Zn2+的浓度,表面极易形成层状(稳定性低、容量高)-类岩盐(稳定性高、容量低)两相共存的表面结构(biphasic surface);基于反应调控,实现两相比例、相变区深度等关键结构特征的精确调控。以此获得了兼具高循环稳定性和高容量的LNMO电极材料

图8 (a–c)随锌含量增加样品表面结构STEM-HAADF图像:(a)尖晶石相和层状相共存;(b)层状相与类岩盐相共存;(c)含锌量高时表面以类岩盐相为主;(d)Zn含量增加LNMO表面相变示意图67Fig. 8 (a–c)STEM-HAADF images to show the surface structure of samples with the increase of Zn content.(a)Coexistence of a spinel phase and a layered phase.(b)Coexistence of the layered phase and the rock-salt like phase. (c)Surface dominated by rock-salt like phase at high Zn content. (d)Schematic illustration for the phase evolution observed on LNMO surface in respond to the change in Zn content 67.
4.2 表面空位调控
在掺杂离子进入表面晶格空位过程中,其除了发生离子取代以外,还有可能进入晶格间隙而发生空位取代。在晶体结构的特定空位上引入特定的金属离子,通过直接作用于过渡金属迁移扩散的通道,能够有效地抑制电化学过程中骨架金属离子自身的迁移,从而减缓对应的结构变化和相转变,实现材料稳定性的有效提升;同时,结合引入金属离子电荷、大小、浓度的系统设计,优化其与附近氧离子的键合,能够进一步增强材料在电化学循环过程中的稳定性。
例如,我们系统考察了AlPO4表面层与LNMO表面晶格的固相反应,发现了Al3+能够进入表面晶格的空位而显著改变LNMO的表面物理化学属性52。实验中,通过在LNMO微米颗粒表面构筑4 nm的AlPO4表面层,控制表面固相反应驱使Al3+渗入LNMO颗粒表面。结合表面的原子结构表征及理论模拟计算,确认了Al3+将优先占据在表面尖晶石晶格的八面体空位(16c)上,该空位是离子扩散的重要通道;通过对扩散通道中Al3+占据比例、占据深度的精确调控,在维持锂离子迁移能力的前提下有效抑制了表面的副反应及锰溶出效果,获得了稳定的电极材料/电解液界面。相关的结构及性能表征如图9所示,针对表面掺杂结构和体相原子结构的球差电镜表征显示表面10 nm深度范围内出现了Al3+,显示出表面固相反应获得可特殊的表面掺杂效果。经过这一表面处理,LNMO在长循环过程中的结构稳定性及电化学稳定性获得了显著提升。
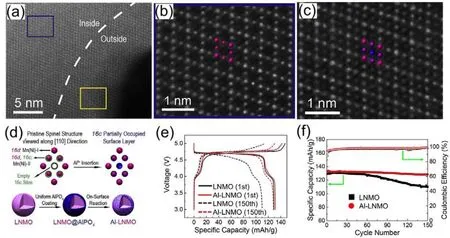
图9 (a)Al-LNMO颗粒的扫描透射电镜-高分辨环形暗场像(STEM-HAADF);(b)和(c)在(a)图中的两个区域的放大的STEM-HAADF图像;(d)表面空位占据(SVSO)策略的示意图;外层大部分的16c位置被外来原子占据;(e)1st和150th的充放电曲线;(f)0.1C倍率条件下的循环性能52Fig. 9 (a)Scanning transmission electron microscopy-high resolution annular dark field image (STEM-HAADF)of Al-LNMO particles; (b)and (c)enlarged STEM-HAADF images of two areas in (a); (d)Surface schematic diagram of the vacancy occupancy (SVSO)strategy; most of the 16c positions in the outer layer are occupied by foreign atoms;(e)The 16c charge and discharge curves at 1st and 150th; (f)Cycle performance at 0.1C rate 52.
鉴于空位在离子传输上的重要作用,在晶体结构的特定空位上引入特定的金属离子,通过直接作用于过渡金属迁移扩散的通道,能够有效地抑制电化学过程中骨架金属离子自身的迁移,从而减缓对应的结构变化和相转变,实现材料稳定性的有效提升;同时,结合引入金属离子电荷、大小、浓度的系统设计,优化其与附近氧离子的键合,能够进一步增强材料在电化学循环过程中的稳定性晶格空位的调控为材料的晶格稳定性提升、离子扩散通道优化等提供了新的机制和手段。在近期的研究中,Guo等人68发现其它的金属离子如Mg2+等也具备选择性占据空位的特性。在其研究中,作者发现基于固相反应引入的Mg2+可以在八面体中心(16c)或是四面体中心(8a)中进行填充,也可以在LNMO中占据空位,从而也可以实现对离子溶出通道的调控,进而显著降低过渡金属的溶出效果,获得LNMO循环稳定性的显著提升。相关研究充分体现了晶格空位调控在控制高比能正极材料表面活性、提升材料应用性能上的有效性和优势。Meng等人69在富锂材料表面20 nm的厚度内,利用表面气相处理的方法成功引入了氧的空位,从而调节了材料表面O的活化程度,使得材料结构稳定性得到了明显的提升,经过100周的循环后材料依然保持300 mAh·g-1的放电容量。
4.3 表面复合调控机制
表面掺杂虽然有助于降低表面的活性,从而对CEI的副反应进行有效控制;但是,鉴于掺杂层仍然具有电化学活性,与完全惰性的物种相比其副反应程度仍然较高,因此,从表面稳定能力的优化出发,控制表面包覆层与正极表面之间的高温固相反应,在促进惰性层中金属离子向正极表面渗入实现掺杂的同时,确保残留部分包覆层在表面,从而获得兼具有表面包覆和表面掺杂效果的复合层,有可能充分发挥惰性物种对正极和电解液的物理隔离效果,在稳定表面晶格的同时获得表面稳定能力的最大化。
例如,Schipper等人70利用ZrO2物种来包覆修饰NMC811材料,其在不同温度下进行表面固相反应以实现NMC材料的表面改性。当处理温度控制在600 °C以下时,ZrO2和NMC811的固相反应程度低,ZrO2包覆层仍在NMC811外层表面显著存在。随着温度升高,固相反应会相应加剧,伴随着ZrO2包覆层的消耗,表面掺杂程度变得明显。作者考察了在800 °C时的表面固相反应效果,确认NMC811表面仍存在未消耗完毕的ZrO2物种(图10a,b),同时NMC自身表面会因为Zr4+的渗入而形成表面掺杂层,这样获得了表面包覆层与表面掺杂层共存的复合调控机制。电化学性能测试确认当两种贡献同时存在时可以使NMC811材料获得最好的循环稳定性,体现了ZrO2表面保护层和Zr4+表面限域掺杂两种作用的共同贡献。

图10 (a)NMC811ZO-800 °C的透射图;(b)表面Zr含量(XPS测试结果)和温度之间的关系;(c)Zr含量与颗粒(NMC811ZO-450 °C和NMC811ZO-800 °C)表面到内部之间的距离之间的关系;(d)未包覆的NMC811和ZrO2包覆的NMC811ZO-450 °C and NMC811ZO-800 °C在30 °C条件下的循环性能70Fig. 10 (a)Transmission diagram of NMC811ZO–800 °C; (b)Relationship between surface Zr content (XPS test result)and temperature; (c)The relationship between Zr content and the distance from the surface of particles(NMC811ZO-450 °C and NMC811ZO-800 °C)to the interior; (d)Cycle performance of uncoated NMC811 and ZrO2 coated NMC811ZO-450 °C and NMC811ZO-800 °C at 30 °C 70.
对于富锂锰基正极材料而言,阴离子(O2-)的氧化还原参与了容量的贡献,同时O在材料结构中起着框架的作用,它的稳定与否直接关系到材料结构的稳定性。通常,富锂材料晶格失氧及结构衰变都是从表面开始发生,因此,如何保持表面晶格中氧的稳定性对富锂材料的电化学性能有着至关重要的作用。单纯的包覆对材料的循环稳定性及倍率性能可以起到较好的作用,但无法有效抑制富锂材料的电压衰减问题。为了实现抑制电压衰减的目的,对富锂材料表面修饰的方法也从简单的包覆向表面混合修饰的策略转变。例如Cho等人71利用联氨处理富锂材料,促使表面发生重构,再通过石墨烯的包覆降低界面的副反应,抑制TM离子的溶出,从而使得材料的循环稳定性及电压衰减都得有了明显的提升。
5 总结与展望
具有高能量密度、高安全性、低成本的锂离子电池技术为促进电动汽车的大规模应用起到了关键推动作用。然而,近年来随着电池能量密度的不断提升,其安全性问题也日益引起研究者的广泛关注,也成为决定电动汽车产业成败的重要因素;相应的,高比能电池体系开发过程中关键电池材料自身的稳定性、安全性的研究也必将成为未来研究的重点和难点。作为下一代锂离子电池高比能正极材料的代表,富镍正极、高电压尖晶石正极以及富锂层状氧化物正极材料等不同材料体系其失效机制及稳定方式的研究也引起了广泛关注。针对不同正极材料自身的物化特性,结合其在电化学循环过程中的稳定性问题,系统研究诸如电极材料腐蚀、阳离子混排、析氧、裂纹产生以及电解液分解等不稳定因素与后果,将是深入认识正极材料失稳机制的重要研究内容;同时,从电极材料的结构调控出发,结合表面纳米结构控制与体相晶格调控等多方面的努力,充分提升下一代高比能正极材料的结构稳定性与电化学稳定性,将成为实现兼具高比能、高稳定性下一代锂离子电池的关键。
对于基于表面限域掺杂的结构调控而言,目前仍存在多方面的挑战。首先,虽然特定掺杂金属离子在表面晶格的引入在电极材料表面物理化学性能的优化设计上显示了积极的效果。但是,鉴于不同电极材料自身晶格结构及掺杂离子与其作用效果的复杂性,目前针对表面掺杂物种的筛选一般采用的是“试错”的方法,基于大量实验来获取既能稳定表面结构又不损害其自身可逆容量掺杂物种。相关的试错过程存在耗时、较为盲目等方面的缺点,而且将掺杂效应进行技术转移,推广到不同体系正极材料上的有效性较低。针对大量正极材料的稳定需求,认知表面修饰材料的作用规律、实现有效掺杂物种的快速筛选具有重要意义。就此而言,结合理论计算的努力展开判断和筛选,将极大地缩小目标掺杂材料的选择范围,为从实验上研究并确认相关的表面掺杂物种提供有力的指导,并极大地加速相关的工作进程。例如,可以充分借鉴材料基因组(MGI)的策略来寻找与确定正极材料表面掺杂离子,通过高通量计算快速获得最优的表面修饰物种与状态,为正极材料表面稳定提供理论指导与智力支持,从而极大加速材料的筛选过程。
其次,不同高比能正极材料自身晶格结构、失稳机制等存在特殊性,对其表面稳定效应的研究往往独立进行且需要较长的研究周期。近年来,随着先进测量技术的发展,在原子尺度上揭示不同表面反应的机理成为可能,也为材料稳定机制的提出提供了可靠的工具和手段。充分结合一系列高精度表征手段的最新进展,针对表面衰减过程的关键过程如过渡金属溶解、晶间裂纹、氧释放、电解质分解等展开研究,结合界面掺杂调控的可靠性评价,实现在原子、分子尺度上获得高比能正极材料的失稳机制和稳定途径也将成为高比能正极的研究热点之一。值得注意的是,诸如环境透射电镜、球差电镜、同步辐射、原位电化学质谱等一系列表征手段已经能够充分应用到电极材料在循环过程中的原位结构表征,为深入理解相关材料的电化学活性和副反应途径提供了特殊途径和有利工具。
最后,表面限域掺杂的实现有赖于在正极材料表面构筑纳米精度均匀包覆层的能力,从电极材料的实际应用需求出发,立足于相关关键工艺的放大,获得大规模处理电极材料表面的能力也将成为高比能正极研究的重点。目前,基于溶液中生长动力学构筑纳米精度包覆层的策略为实现粉体的表面精确调控提供了可能的解决方案,但如何实现从实验室量级的材料制备向工业级别生产的跨越仍需在合成方法、工艺放大上开展系统研究,亟需探索适应于工业生产需要的合成方法,从而获得对大量粉体表面处理的同时确保表面纳米结构控制的有效性和精确性。此外,电极材料在液相处理过程中自身稳定性的变化也是值得关注的问题。例如,对于下一代高镍三元正极材料而言,其对水汽敏感,两者接触会促使高镍材料结构发生坍塌而造成电化学活性损失。因此,基于水相的液相包覆途径本身会对高镍材料造成严重损害,进而也使得基于水相的表面控制途径无法有效应用于高镍三元等正极材料。可以预见的是,从正极材料自身特性出发,开发基于非水溶剂体系的表面控制路线将成为高镍三元材料等研究的重要内容。
