儿童X-连锁肾上腺脑白质营养不良39例临床特征分析
2021-11-18韦紫倩林晓滨李淑梅蔡燕娜邵咏贤梅慧芬郑锐丹梁翠丽刘鸿圣江敏妍
韦紫倩 林晓滨 李淑梅 蔡燕娜 邵咏贤 程 静 梅慧芬 郑锐丹 梁翠丽 刘鸿圣 江 华 江敏妍 刘 丽
广州医科大学附属广州市妇女儿童医疗中心(广东广州 510623)
X-连锁肾上腺脑白质营养不良(X-linked adre-noleukodystrophy,X-ALD)是一种由过氧化物酶体遗传缺陷引起的脂质代谢性疾病,呈X-连锁隐性遗传。编码过氧化物酶膜蛋白(ATP binding cassette subfamily D member 1,ABCD 1)基因ABCD 1位于Xq28[1],其发生变异时,肾上腺脑白质营养不良蛋白(adrenoleukodystrophy protein,ALDP)生成缺陷,无法将极长链脂肪酸(very long-chain fatty acids,VLCFAs)转运进过氧化物酶内进行β氧化,从而在脑白质、肾上腺皮质等组织器官中堆积[2],引起脑白质脱髓鞘、肾上腺功能不全等病理变化。本文回顾分析39例X-ALD患儿的临床资料,分析其临床特征、治疗及预后。
1 对象和方法
1.1 研究对象
回顾分析2011年至2018年广州市妇女儿童医疗中心确诊的X-ALD 患儿的临床资料。研究对象入选标准:①<18岁;②符合X-ALD诊断标准;③获取家长和/或患儿知情同意。排除标准:①血浆VLCFAs未见升高;②行ABCD 1基因变异分析为阴性者;③X-ALD女性携带者。
X-ALD诊断依据[3-4]:①进行性神经系统受累症状,如步态不稳、认知障碍、视力下降等;②肾上腺皮质功能减退症,如皮肤色素沉着、皮质醇水平降低、促肾上腺皮质激素(ACTH)水平升高;③血浆VLCFAs异常升高;④头颅磁共振成像(MRI)显示脑白质脱髓鞘病变;⑤基因分析提示ABCD1基因存在致病变异;⑥符合X-连锁隐型遗传模式。其中,符合①~⑥者确诊为儿童脑型X-ALD,符合②③⑤⑥者确诊为单纯肾上腺型X-ALD。根据致病基因ABCD1碱基变异类型,将儿童脑型X-ALD患者分为碱基替换组及缺失、重复组两组。
1.2 方法
1.2.1 临床资料收集 收集患儿的发病年龄、确诊年龄、临床表现、实验室检查、头颅MRI检查、ABCD1基因变异分析、治疗与随访等资料。
1.2.2 实验室检查 对疑似X-ALD 患儿进行检查的项目包括血浆VLCFAs、血浆皮质醇、ACTH 和血气、电解质。血浆VLCFAs包括二十六烷酸(C26:0)、二十四烷酸(C24:0)和二十二烷酸(C22:0),另外还计算C22:0与C26:0比值(C26:0/C22:0)和C24:0与C22:0比值(C24:0/C22:0),并将其作为筛查指标。VLCFAs 采用同位素标记气相色谱-质谱联用定量法(gas chromatographic mass spectrometric,GC-MS)检测,血浆皮质醇、ACTH由化学发光分析仪测量。
1.2.3 基因分析 采集患儿外周血2 mL,采用聚合酶链式反应(PCR)扩增ABCD 1基因编码区,利用ABI 3730 DNA分析仪对PCR产物进行直接测序,所得结果与NCBI 上的ABCD 1序列进行比对,并利用Polymorphism Phenotyping v2(PolyPhen-2,http://genetics.bwh.harvard.edu/pph 2/)分析新发变异的 致病性。1.2.4 Loes 评分 根据1994 年Loes 等[5]针对脑型X-ALD 患者建立的Loes 评分,对头颅MRI 白质病变累及部位,如胼胝体、脑桥、丘脑、枕叶、额叶等进行疾病严重程度评分。采用Loes评分对脑型患儿大脑的萎缩程度、白质病变范围以及位置进行评估。
1.3 统计学分析
采用SPSS 21.0统计软件进行数据分析。符合正态分布的计量资料以均数±标准差表示;非正态分布计量资料以中位数(四分位数范围)表示,两组间比较采用Wilcoxon秩和检验。以P<0.05为差异有统计学意义。
2 结果
2.1 一般情况
纳入39例患儿,其中儿童脑型X-ALD 27例,单纯肾上腺型X-ALD 12例。中位发病年龄6.0(4.0~7.0)岁,中位确诊年龄6.0(5.0~8.0)岁。儿童脑型X-ALD中位发病年龄6.0(5.3~7.8)岁,中位确诊年龄7.0(6.0~8.0)岁;单纯肾上腺型X-ALD中位发病年龄3.0(1.0~6.0)岁,中位确诊年龄5.0(2.8~7.5)岁。其中2例单纯肾上腺型X-ALD患儿经过数年演变成儿童脑型X-ALD。
2.2 临床表现
27例儿童脑型X-ALD 患儿主要表现为:①运动功能障碍,包括步态异常17例、抽搐7例、肢体乏力3例、运动发育迟缓1例;②认知功能损害,表现为学习成绩下降、记忆力下降、注意力不集中、反应迟钝的智力减退16例,性格改变8例;③视听功能损害和语言障碍,包括视力下降或视物模糊15例、听力下降9例、书写困难5例,表现为语速减慢、吐字不清等语言表达能力下降12例。12例单纯肾上腺型X-ALD患儿,10例表现为皮肤色素沉着,2例无明显临床表现。27例儿童脑型患儿首发与确诊时的临床症状有变化,其中有步态异常患儿由33.3%增至63.0%,智力减退者由37.5%增至59.3%,视力下降或视物模糊者由37.5%增至55.6%,语言表达能力下降者由16.7%增至44.4%。此外有听力下降、皮肤色素沉着、性格改变、抽搐、书写困难、肢体乏力者均增加。
2.3 实验室检查
39例X-ALD患儿C26:0水平均升高,20例C24:0升高;所有患儿C26:0/C22:0和C 24:0/C 22:0 均较正常值升高。27例儿童脑型及2例单纯肾上腺型演化为儿童脑型患儿C 26:0/C 22:0 中位数 0.07 μmol/L(正常值<0.013 μmol/L),C 24:0/C22:0中位数2.15 μmol/L(正常值<1.04 μmol/L)。
2.4 影像学检查
27例儿童脑型X-ALD 患儿均有脑白质受累,头颅MRI表现为受累部位T2WI呈高信号,病变区域为对称性,主要累及胼胝体、幕上白质区(包括额、顶、枕、颞叶)、视觉通路,听觉通路、投射纤维所在区域和小脑。12例单纯肾上腺型患儿,除2例演化为儿童脑型者外,其余无头颅影像学改变。由影像科医师对患儿进行Loes评分[5]。在脑型(含2例由肾上腺型演化者)患儿中,Loes评分9分以下25例,9分以上3例,1例患儿影像学检查结果缺如。
2.5 基因检测
39例X-ALD患儿均行ABCD1基因检测。在29例脑型(含2例由肾上腺型演化者)患儿中,碱基替换17例,缺失11例,重复1例;在10例单纯肾上腺型患儿中,碱基替换5例,缺失3例,插入2例。所有患儿中发现3 个新发变异,分别为c.578C>G(p.Prp193Arg)、c.1615 A>C(p.Met 539 Leu*)和c.2_5 dupTGCC(p.Pro 2=fs*),均未见报道,未被X-ALD 数据库(www.x-ald.nl)收录。通过Polyphen 2 软件预测,c.578C>G和c.1615A>C很可能致蛋白质功能受损;c.2_5dupTGCC可使终止密码子提前出现,形成截短蛋白,影响ALDP蛋白质合成,推测可能致病。
2.6 基因型与临床表型分析
在脑型患儿(含2例由肾上腺型演化者)中,18例为碱基替换组(其中1例影像学检查结果缺失),Loes评分为6.00(4.00~7.75)分;11例为缺失、重复组,Loes 评分3.00(1.00~5.00)分,两组间Loes 评分差异有统计学意义(Z=2.19,P=0.028),碱基替换组患儿的Loes评分高于缺失、重复组。
2.7 治疗及预后
39例患儿均给予氢化可的松替代治疗,2例出现继发性癫痫给予安定、卡马西平等对症处理,5例行异基因造血干细胞移植(allogenic hematopoictic stem cell transplantation,HSCT)治疗。对患儿的随访包括间隔6个月的定期复诊和电话追踪,截至2020年10月。9例患儿死亡,其中2例于行HSCT后死亡;30例存活。接受HSCT 治疗的患儿中,3例术后病情平稳,2例于术后2个月和1年死亡;4例移植术后血浆 VLCFA 较前下降,但未恢复至正常水平,2例神经系统症状较术前有加重,2例病情平稳。见表1。
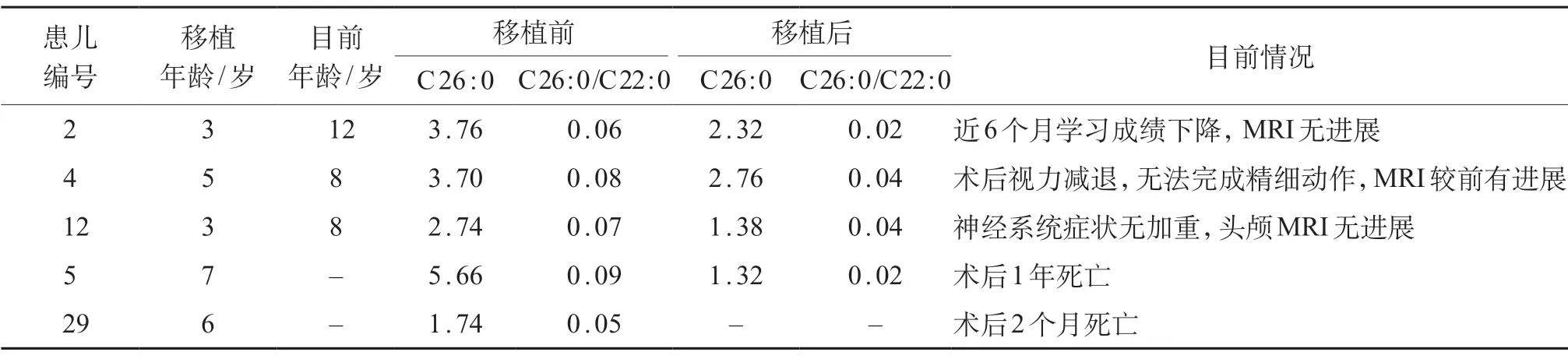
表1 脑型患儿移植前后情况
3 讨论
根据发病年龄、临床表现,X-ALD分为7种类型,包括儿童脑型、青少年脑型、成人脑型、肾上腺脊髓神经病型(adrenomyeloneuropathy,AMN)、单纯肾上腺型、无症状型以及女性杂合子。本研究39例患儿均为男童,首诊时有27例为儿童脑型X-ALD,12例为单纯肾上腺型X-ALD。单纯肾上腺型X-ALD患儿间隔3~6个月检查头颅MRI,其中2例于确诊后1年和3年出现不同程度神经系统受累,由单纯肾上腺型X-ALD向儿童脑型X-ALD转变。儿童脑型X-ALD平均发病年龄与平均确诊年龄相近(仅相差4个月);单纯肾上腺型X-ALD中位发病年龄与中位确诊年龄相差2年,提示该临床表型起病隐匿,容易被忽视。这是因为儿童脑型X-ALD常以认知、视力、运动障碍为首发表现,表现为学习成绩下降、注意力不集中、口齿不清、走路步态异常,甚至出现反复抽搐,引起家长重视而就医;但单纯肾上腺型X-ALD 仅以皮肤黏膜色素沉着为主要表现,部分患儿曾因此至皮肤科就诊。一旦发生神经系统病变,病程即呈进行性发展,本研究儿童脑型X-ALD首发症状和确诊时临床表现存在差异,在短时间内神经系统受累症状均有不同程度加重,表明儿童脑型X-ALD病情进展迅速。
因X-ALD 的临床症状不典型,且进展速度快,因此临床医师需提高警惕,对出现类似症状者作排除性诊断[6]。对于皮肤色素沉着者,或出现视、听觉损害,认知、运动障碍者,需仔细行神经系统体检及皮质醇、ACTH、VLCFAs、头颅影像学检查。ABCD1基因变异导致ALDP 缺陷,使VLCFAs 不能转运进入过氧化物酶体进行β-氧化进一步分解代谢,故细胞和血浆内VLCFAs 增高,尤以C 24:0 和C 26:0增高明显。因此,脑型X-ALD 患者头颅MRI 表现较典型,大多表现为胼胝体压部、幕上白质区、视通路的对称性脱髓鞘改变,而单纯肾上腺型X-ALD 脑部无脱髓鞘改变,但应每3 个月复查头颅MRI,以早期识别单纯肾上腺型X-ALD 向脑型X-ALD 转变 可能[3]。
ABCD1基因分析为确诊X-ALD的金标准[7]。本研究39例X-ALD 患儿均发现基因变异,同时发现3个新发变异。c.578 C>G(p.Pro 193 Arg),造成第193号位的非极性氨基酸脯氨酸变为极性氨基酸精氨酸,氨基酸性质改变影响过氧化物酶ALDP的功能,导致VLCFAs堆积[7];c.1615A>C(p.Met539Leu*)变异,造成539 号蛋氨酸被亮氨酸取代,蛋氨酸可以通过S腺苷甲硫氨酸提供活性甲基,减少ALDP的活性甲基从而致病;c.2_5dupTGCC(p.Pro2=fs*)移码变异可使终止密码子提前出现,形成截短蛋白,推测截短蛋白将降低ALDP 酶活性[8],但需进一步功能学验证。此外,由于X-ALD为X-连锁隐性遗传,因此对于确诊患者,若有同胞兄弟,需积极筛查,同时对患儿父母提供遗传咨询[9]。
既往文献报道,不同碱基变异类型的特征性代谢产物C26:0水平有统计学差异,插入或缺失变异组的C 26:0 水平显著高于碱基替换组[10]。本研究分析了ABCD 1基因变异类型和Loes 评分之间的关系。变异类型为碱基替换的儿童脑型X-ALD 患儿,发病和确诊年龄均比缺失、重复组晚,Loes 评分亦高。可以推测,由于非碱基替换组的变异大多导致终止密码子提前出现,形成截短蛋白,后者可能残存部分ALDP 活性,因此神经系统受损相对较轻;同时由于缺失、重复组的发病年龄相对较早,比碱基替换组患儿发病提早1年,最终导致Loes评分相对较低,存在HSCT的适应证。
X-ALD治疗主要为对症治疗和HSCT[11]。对于单纯肾上腺型X-ALD,由于存在肾上腺皮质功能低下,可予氢化可的松替代治疗;早期儿童脑型X-ALD,即Loes评分低于9分者,可行HSCT治疗。随访研究发现,早期HSCT可降低血液及脑组织中VLCFA浓度,延缓病情进展,提高X-ALD患儿生存率及生活质量。但中晚期儿童脑型X-ALD 以及脊髓神经病型X-ALD,目前尚无有效方法阻止病情进展[12-14]。
