COL6A1内含子变异致Ullrich先天性肌营养不良1例报告并文献复习
2021-11-18林明星邱鸣琦吴传军陈燕惠
胡 君 林明星 邱鸣琦 吴传军 陈 铮 陈燕惠
福建省临床重点专科 福建医科大学附属协和医院儿科(福建福州 350001)
Ullrich 先天性肌营养不良(Ullrich congenital muscular dystrophy,UCMD)是Ⅵ型胶原蛋白相关性肌病之一,由COL 6基因(COL 6 A 1,COL 6 A 2和COL 6 A 3)变异引起Ⅵ型胶原蛋白功能缺陷,导致以肌无力、近端关节挛缩、远端关节活动过度为主要临床表现的遗传性肌病[1]。UCMD以新发变异导致的常染色体显性遗传为主,亦可见常染色体隐性遗传[2]。其患病率约为1.3/1000000[3],已纳入国家罕见病目录。COL 6基因内含子变异所致Ⅵ型胶原蛋白相关性肌病的国内报道罕见。现报告1例COL6A1内含子变异(+189 C>T)所致UCMD的临床表型及遗传学分析,并复习相关文献。
1 临床资料
患儿,女,4岁,因运动发育落后于2019年1月来福建医科大学附属协和医院就诊。患儿2岁时容易摔跤,下蹲后站起困难,上下楼梯需要人扶,不会跑、跳;认知发育未发现异常。患儿系 G2P2,足月剖宫产,生后无窒息、抢救史,母孕期体健。患儿4 月龄竖头,6月龄翻身,8月龄独坐,不会爬,13月龄扶站,16月龄独立行走。父母非近亲结婚,家族无类似病史。患儿哥哥9岁,身体健康。体格检查:体质量15.6 kg(P75)、身长103.5 cm(P50)、呼吸22次/min、心率83次/min、血压95/60 mmHg;神清,应答如常,圆脸,手心、足心皮肤柔嫩,心肺无异常;双上肢近端肌力Ⅳ级,远端肌力Ⅴ级,双下肢近端肌力 Ⅳ 级,远端肌力Ⅳ 级;四肢肌张力稍减低;四肢关节活动过度,双膝、踝关节挛缩,右侧更为显著;脊柱前凸,双足跟骨后凸;双侧跟、膝腱反射均未引出,双腓肠肌无肥大;病理征阴性。见图1。实验室检查:血常规无异常;丙氨酸转氨酶 25 U/L,天冬氨酸转氨酶40 U/L,乳酸脱氢酶293 U/L,磷酸肌酸激酶285 U/L,磷酸肌酸激酶同工酶41 U/L。肌电图:肌源性损害改变为主(上下肢近端肌),伴轻度神经源性损害(上下肢远端肌)。
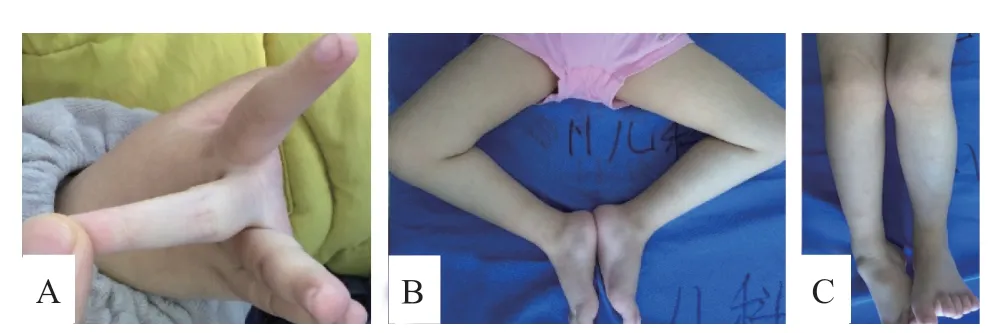
图1 UCMD 患儿四肢外观特征
根据患儿临床表现及辅助检查结果,拟诊先天性肌营养不良。为进一步明确诊断,经患儿家长知情同意及医学伦理委员会批准(No.2020 KY 023),取患儿及其父母外周静脉血各2 mL,行全外显子测序(whole exome sequencing,WES)联合“加密”技术检测(福君基因公司),并行Sanger验证。结果显示,患儿COL 6 A 1基因(NM_001848.2)存在内含子新发变异(c.930+189 C>T)。Sanger 验证显示,患儿父母未见变异,为野生型。见图2。该变异为已知致病变 异[4]。综合患儿临床表现、相关实验室检查,明确诊断为UCMD。随后,对患儿进行肌肉按摩、康复训练等治疗。患儿随访至今仍可独自步行,四肢肌力、肌张力与就诊前相仿,无明显呼吸功能不全,脊柱前凸角度较小,无需夜间无创通气呼吸支持和脊柱矫形治疗。

图2 患儿及父母COL6A1基因Sanger 测序图
2 讨论
UCMD 发病较早,通常出生后即发病,表现为肌无力和肌张力低下,运动发育延迟,并以近端关节挛缩、远端关节活动过度为特点[1,5-8]。UCMD常合并先天性斜颈、先天性髋关节脱位、脊柱侧弯、跟骨后凸等;其他特征包括圆脸、下眼睑下垂、耳朵突出、皮肤毛囊过度角化、瘢痕体质等。UCMD患儿多在10~ 20岁因呼吸衰竭而死亡。UCMD患儿的肌酶正常或轻度升高;肌电图表现为近端肌的肌源性损害,或远端肌的神经性损害或无异常[1,5-8]。
本例患儿2岁起病,表现为肌无力和肌张力低下,运动发育延迟,容易摔跤,下蹲后站起困难,远端关节活动过度;圆脸,脊柱前凸,双足跟骨后凸;肌酶轻度升高;肌电图示肌源性损害(上下肢近端肌),伴轻度神经源性损害(上下肢远端肌)。患儿表现部分符合 UCMD 的典型临床表现。为进一步明确诊断,行WES基因检测加基因拷贝数变异检测,初步结果为阴性。于是在此基础上做“加密”,即除覆盖上述的基因区域以外,对存在已知致病变异的基因内含子区、调控区等特殊区域设计更多额外的覆盖探针,从而捕获到1个低覆盖区的内含子新发变异位点COL6A1基因NM_001848.2:c.930+189 C>T,为已知致病变异[2]。因此,结合临床表现及相关检查结果,本例患儿符合UCMD诊断。由于患儿家属拒绝行肌肉MRI、肌肉活检,致使诊断依据有所欠缺。
以“congenital muscular dystrophy,collagen VIlike dystrophy,Ullrich congenital muscular dystrophy,Bethlem myopathy,COL 6 A 1,intron”为关键词检索HGMD 和PubMed 文献数据库,并以“先天性肌营养不良、Ⅵ型胶原蛋白相关性肌病、Ullrich 先天性肌营养不良、Bethlem 病、COL 6 A 1、内含子”为关键词检索中国知网、维普、万方等中文文献数据库,检索时间从建库至2020 年12 月。检索到相关文献4 篇,涉及COL6A1+189 C>T基因变异的患儿35例[2,4,9-10]。加上本例患儿,共36例UCMD 患儿,男16例、女20例。36例患儿临床表现与UCMD 的典型临床表现有所区别,即症状出现延迟,随后加速进展。大多数患儿在新生儿期症状相对较少,婴幼儿期表现为肌无力和肌张力低下,运动发育延迟。随着年龄增长,逐渐出现UCMD 典型临床表现,36例患儿中位起病年龄 4.2(0.5~9.5)岁,首发症状以易摔跤,上下楼梯困难多见;中位年龄9.0(3.0~14.0)岁时失去行走能力,需要全程使用轮椅;中位年龄13.0(7.0~21.0)岁时出现呼吸功能不全,需要夜间无创通气支持;同时,脊柱侧弯、近端关节挛缩、远端关节活动过度等表现加速进展。
COL6A1+189 C>T基因新发变异所致UCMD的临床表现与UCMD的典型临床表现有所区别的原因,可能与COL6A1基因的框内假外显子(内含子)插入变异机制相关[10]。考虑为:①假外显子剪接位点(变异的剪接供体和隐性剪接受体)的外显率不完全,使插入假外显子的转录本被稀释,并可能受到时间调控;②由含α1链的变异肽形成负显性或有害变异模式;③两者兼而有之。假外显子编码位于三螺旋结构域(重复的 Gly-X-Y 三联体亚基)N 端具有潜在破坏性的非胶原肽,破坏了对二聚体稳定至关重要的半胱氨酸残基。同时,含α 1 链的变异肽将假外显子掺入 Ⅵ 型胶原蛋白中,使得四聚体形成串珠状微纤维受阻,形成不规则的基质。此外,假外显子编码的非胶原肽富含带正电的残基通过错误折叠的结构破坏三螺旋结构域。通过以上步骤,导致 Ⅵ 型胶原蛋白的组装和功能遭到全面破坏。
需要注意的是,UCMD的肌电图表现出可变的特征,近端肌为肌源性损害,远端肌伴或不伴神经源性损害,这与肌肉损伤后自我修复的过程相关,与基因型或表型无明确的对应关系[7-8,11]。在UCMD肌肉损伤后修复、再生过程中,肌电图可出现宽时限的多相运动单位电位(motor unit potentials,MUPs),但募集减少;或MUP晚成分和原来的主波相隔较远,呈现出神经源性损害表现。此外,若记录电极尖端靠近再生肥大的肌纤维,会出现高波幅MUP,类似神经源性损害,但在主尖峰内的面积很小。因此,肌电图检查结果可作为UCMD诊断的参考依据,但不能作为排除依据。
UCMD目前没有特异性治疗方法,主要是对症支持治疗[12]。如出现呼吸困难时,予以呼吸支持,从而有效减轻其症状和改善生活质量。对于脊柱侧弯,可使用站立架、定位支撑和脊柱外科手术方法,尽管这些方法的有效性存在争议。COL6A1+189 C>T变异的发现为UCMD提供了两种潜在的基因治疗方法[2,10]。利用Gapmer反义寡核苷酸(antisense oligonucleotides,ASO)激活核糖核酸酶H,特异性地沉默含α1链负显性变异的RNA转录产物,促进这种异常剪接的外显子跳跃(或假外显子跳跃)。另一种方法是使用ASO 干扰 pre-mRNA,以剪接COL6A1+189 C>T变异的假外显子,实现外显子跳跃(或假外显子跳跃),生成野生型COL6A1转录产物。本例患儿明确诊断后行肌肉按摩、康复训练等治疗,随访至今仍可独自步行,四肢肌力、肌张力与就诊前相仿,无明显呼吸功能不全,脊柱前凸角度较小,无需夜间无创通气呼吸支持和脊柱矫形治疗。
UCMD 以新发变异导致的常染色体显性遗传为主[2]。尽管COL6A1新发变异的患儿同胞再发风险较低(1%),但不排除其父母的外显率降低和/或胚系嵌合存在的可能性[13-14]。若患儿家庭想再次生育,应进行遗传咨询和产前诊断。
综上所述,COL 6 A 1+189 C>T 新发变异所致UCMD的临床表现与UCMD典型临床表现有所区别,即症状出现延迟,随后加速进展。UCMD 肌电图检查结果可变性大,不能作为排除本病的依据;基因检测有助于诊断。UCMD 目前没有特异性治疗方法,遗传咨询和产前诊断尤为重要。
