融合多视角和多标签学习的RNA 结合蛋白识别
2021-11-17杨海涛邓赵红王士同
杨海涛,邓赵红,王士同
江南大学 人工智能与计算机学院,江苏 无锡214122
核糖核酸(ribonucleic acid,RNA),存在于生物细胞以及部分病毒、类病毒的遗传信息载体之中,在生命体中主要发挥调控编码基因表达的作用,同时也担任基因转录后合成蛋白质的模板角色,是生命体中不可缺少的成分。一条RNA想要顺利发挥其功能,一般需要借助RNA 结合蛋白(RNA-binding protein,RBP)进行介导,因此缺少某种RBP 可能会导致某类RNA 无法发挥其调控或翻译的功能,使生命体的某些重要蛋白质缺失或异常增殖,影响自身机能。
RNA 结合蛋白(RBP)是翻译过程的关键参与者,它们结构域的多功能性与结构灵活性使得RBP能够控制大量转录物的代谢。RBP 几乎涉及翻译调控层的所有步骤,它们与其他蛋白质以及编码和非编码RNA 建立高度动态的相互作用,产生称为核糖核蛋白复合物的功能单元,调节RNA 剪切、多腺苷酸化、稳定性、定位、翻译和退化[1-2]。研究发现,某些特定RBP 具有调节RNA 合成癌蛋白和肿瘤抑制蛋白的功效,因此破译RBP 与癌症相关RNA 靶标之间错综复杂的结合关系网络将为肿瘤生物学提供更好的研究方向,并可能发现治疗癌症的新方法[3-4]。
在大数据和测序技术高度发展的背景下,医疗条件无法对每对RNA 和RBP 进行结合性检测,因此涌现了很多利用机器学习模型从RNA 序列中识别RBP 结合位点[5]的算法。例如:Maticzka 等人提出了GraphProt[6]方法,其从高通量实验数据中学习RBP序列和结构的结合偏好,设计出独特的计算框架;Corrado 等人提出RNACommender[7],一种预测结合位点的方法,能够通过可用的相互作用信息,考虑蛋白质结构和RNA 的模拟二级结构,向未探索的RBP推荐RNA 靶点;由Zhang 等人提出的HOCNNLB[8]使用高阶核苷酸编码来作为初始特征,预测某段给定的RNA 是否是结合位点。这些方法的关注点在于利用原始RNA 序列的特征预测结合位点[9-10],忽视了RNA 与RBP 已有的结合信息对预测的助力。针对此,Pan 等人提出了iDeepM[11]方法,其利用多标签分类和深度学习法预测一条RNA 与多种RBP 的结合情况,成功达到多标签分类的预期效果。但iDeepM也存在如下的不足:其使用的RNA 序列单视角数据虽然对分类具有一定的有效性,但受限于RNA 序列的信息量不足,导致预测精度较低;另外该方法使用的卷积神经网络和长短时记忆网络未能充分学习到标签之间的关联,同样对预测精度产生影响。
本文针对iDeepM方法面临的挑战,在Pan等人工作的基础上进行了改进,提出了RRMVL(RNA-RBP multiview learning)方法。RRMVL 融合了多视角深度特征学习、多标签特征学习和最优多标签链式学习技术来进行RBP 识别。首先,在最初的RNA 序列数据基础上,提取了氨基酸序列视角数据、多间隙二肽成分视角数据和RNA 序列语义视角数据;然后,针对不同视角的数据结构,设计各自视角的深度神经网络模型进行深度特征学习;接着,融合提取到的各个视角的深度特征,使用逻辑回归原理学习每个视角的每一维特征对每一个标签的贡献权重;最后,将深度特征向量与各自标签对应的权重系数相乘,输入至改进后的CC 多标签分类器中训练,实现最优多标签链式学习,进一步提高RNA 与RBP 结合的预测精度。本文的实验研究表明使用基于多视角的最优多标签链式学习的方法在预测精度方面有了明显的提升。
本文主要贡献可归纳如下:(1)使用Word2Vec 技术训练6 聚体RNA 语义模型,提取出RNA 序列的序列语义视角数据用于识别RNA 结合蛋白;(2)使用多间隙二肽成分表示法构建RNA 序列的成分视角,其包含更丰富的成分信息;(3)设计独立的网络模型,学习本文提出的RNA 序列视角、氨基酸序列视角、多间隙二肽成分视角和RNA 序列语义视角中每一维深度特征对每个标签的贡献权重,实现多标签特征学习;(4)改进现有的CC 多标签链式分类器,将多标签特征学习后的加权特征向量应用于多标签学习中,最大化提升了CC 多标签分类器对每个标签的学习能力,达到最优多标签链式学习的效果;(5)研究了类样本不均衡对预测RNA 和RBP 结合精度的影响。
1 相关工作
1.1 RNA 序列与氨基酸序列的相互转化
自然界中存在五种碱基A、C、G、U、T,其中前四者是构成RNA 的主要成分。RNA 序列和氨基酸序列可以通过翻译和逆翻译机制[12]相互转化。因为氨基酸序列是由20 种氨基酸构成的蕴含一定上下文信息的序列,其信息量远比RNA 序列丰富[13],因此在以RNA 为研究主体的生物信息学领域,通常将RNA 序列转化为氨基酸序列进行特征提取与分析。将RNA序列翻译成氨基酸序列是单向且唯一的,但是由于一种氨基酸可对应多种碱基组合,用普通的翻译方法得到的氨基酸序列无法还原至原始RNA 序列,这会造成信息丢失和信息曲解的后果。例如碱基组合GCA 可翻译得到固定的氨基酸A,但是氨基酸A 却可以表示为GCA、GCC、GCG、GCU。为了处理这个问题,本文使用三种方式将RNA 序列翻译为氨基酸序列:(1)从头开始翻译RNA 序列;(2)跳过RNA 序列第一个碱基开始翻译;(3)跳过RNA 序列第一和第二个碱基开始翻译。用此方法可将长度为m的RNA序列转化为3 条长度为1/3m的氨基酸序列,这三种形态的氨基酸序列可以通过序列信息互补还原原始RNA 序列信息。如上述的碱基组合GCA,可使用三种形态序列对应位置的氨基酸R、A、H 来唯一确定。三种形态拼接起来的长度为m的氨基酸序列能够完全继承原始RNA 序列的序列信息,且具有更加丰富的表现形式。
1.2 多间隙二肽成分表示法
二肽是氨基酸序列特有的一种结构[14],任意两个氨基酸的组合称为二肽。因为二肽对左右氨基酸的排列敏感[15-16],所以20 种天然氨基酸可以组成400 种不同的二肽。多间隙二肽成分表示法(g-gap dipeptide composition)[17]是一种描述氨基酸序列中二肽成分信息的方法。这种方法不仅包含了两个氨基酸在序列上的相关性,还描述了由于蛋白质二级结构中的氢键作用,序列上距离远的两个氨基酸,在三维空间上却可能相邻。因此使用多间隙二肽成分表示法可以为机器学习提供更多氨基酸序列和RNA 序列的成分信息,通常将氨基酸序列中多间隙二肽的种类及数量映射为一条特征向量作为初始特征使用。多间隙(g-gap)中的g表示某种中间间隔了g个氨基酸的二肽,取值范围为0 到9。
1.3 基于Word2Vec方法的语义模型
在大数据为背景的信息时代,涌现出众多形式的信息,其中文本信息是最传统也是信息量最大的一种表现形式。如何将文字量化为可以提取特征的数字形式,即自然语言处理(natural language processing,NLP),成为机器学习领域一个重要的研究方向。Onehot 是目前较为流行的编码技术[18-19],其原理是将由n种元素组成的长度为m的文字序列构建为n×m的矩阵,其中把每种元素转化为n维标准正交基向量填充至m长度中的对应位置。但这种方法构建的初始特征矩阵受限于维度过大,且使用机器学习方法提取稀疏矩阵特征的效果不理想,因此one-hot 并不适用于大型词库的文本处理。
Word2Vec[20-22]是NLP 领域常用的方法,其原理是通过训练独特的网络模型,将词库中的每个词映射为k维实数向量,使用词的实数向量来构建文本样本的初始特征矩阵。词向量模型的训练过程如下:(1)对文本样本进行分词操作,构造词典,统计词频,依照词语出现概率构造哈夫曼树,生成每个词语的二进制码;(2)构建一个3 层结构的网络模型,将相邻词语的二进制码分别作为特征和标签输入至模型进行训练;(3)获取模型隐含层参数,计算词库中每个单词的词向量。基于词频的哈夫曼编码可以让词频相似的词在隐藏层激活的内容基本一致,且单词出现的频率越高,它们激活的隐藏层节点数目就越少,有效通过较低的计算复杂度学习到单词在高维空间中的距离分布。通过此方法训练出来的词向量不仅具有维度低的优点,而且包含了其在文本样本中的上下文信息,可以为特征提取提供良好的帮助。
1.4 多标签学习
不同于多分类问题,多标签分类[23]是一种更普遍且更具有挑战的问题,它描述了一个样本可以对应多个类的情况。现处理这类问题的方法有两种:一种是问题转化法,即把多标签问题中的多个标签通过一定形式的组合,变为若干标签集合,将标签集合看作特殊的标签,间接把问题转化为普通的单标签学习问题。经典的算法有BR(binary relevance)[24]、LP(label powerset)[25]和CC(classifier chains)[26]。BR 算法设计若干分类器,有效学习到每个类别的特征,却忽略了标签之间的相关性;LP 算法虽然考虑到标签之间的联系,但是该算法的时间和空间复杂度比较高;CC 算法利用多个分类器构造链式结构,可以有效学习到标签之间错综复杂的关系。另一种做法是改进现有的单标签学习法来适应多标签分类,使其具有处理多标签问题的能力。比较常见的有基于Boosting的算法AMH(AdaBoost.MH)和AMR(AdaBoost.MR)[27],以及基于决策树的算法。其中AMH 是以Hamming Loss作为损失函数来构建学习模型;AMR算法以Ranking Loss 作为损失函数;Clare 等人对经典的单标签决策树学习模型进行了改进,提出算法C4.5[28],其原理是通过计算训练样本的信息增益来训练分类器。改进后的算法中,叶节点不再是一个类,而是一个标签集合。但这些算法没有充分考虑标签间的关联性。
2 多视角多标签识别RNA 结合蛋白
2.1 总览
本文把探索一条RNA 与多种RNA 结合蛋白(RBP)结合的问题转化为机器学习中的多标签分类问题。RNA 序列作为研究主体,RBP 作为类别。不同于现有方法,本文利用分子生物学原理,把原始RNA 序列转化为氨基酸序列,统计氨基酸序列的0-gap 二肽和1-gap 二肽数量,组成多间隙二肽成分,利用Word2Vec 技术构建6 聚体RNA 词向量,由此得到RNA 序列视角、氨基酸序列视角、多间隙二肽成分视角和RNA 序列语义视角的初始数据。然后使用深度卷积神经网络(convolutional neural network,CNN)分别提取4 个视角的深度特征,将其拼接并投入至多标签特征学习模型中训练,通过此模型的处理,可以获取每个标签相关的加权特征向量。接着将加权特征向量投入多标签分类器CC 模型学习标签之间的关联性。最后使用上述CC 模型训练出来的分类器,预测一条未探索的RNA 序列与多种RBP 的结合情况。本文方法的整体框架如图1 所示,它包含4 个部分:获取初始多视角数据、多视角深度特征学习、多标签特征学习和多标签学习。
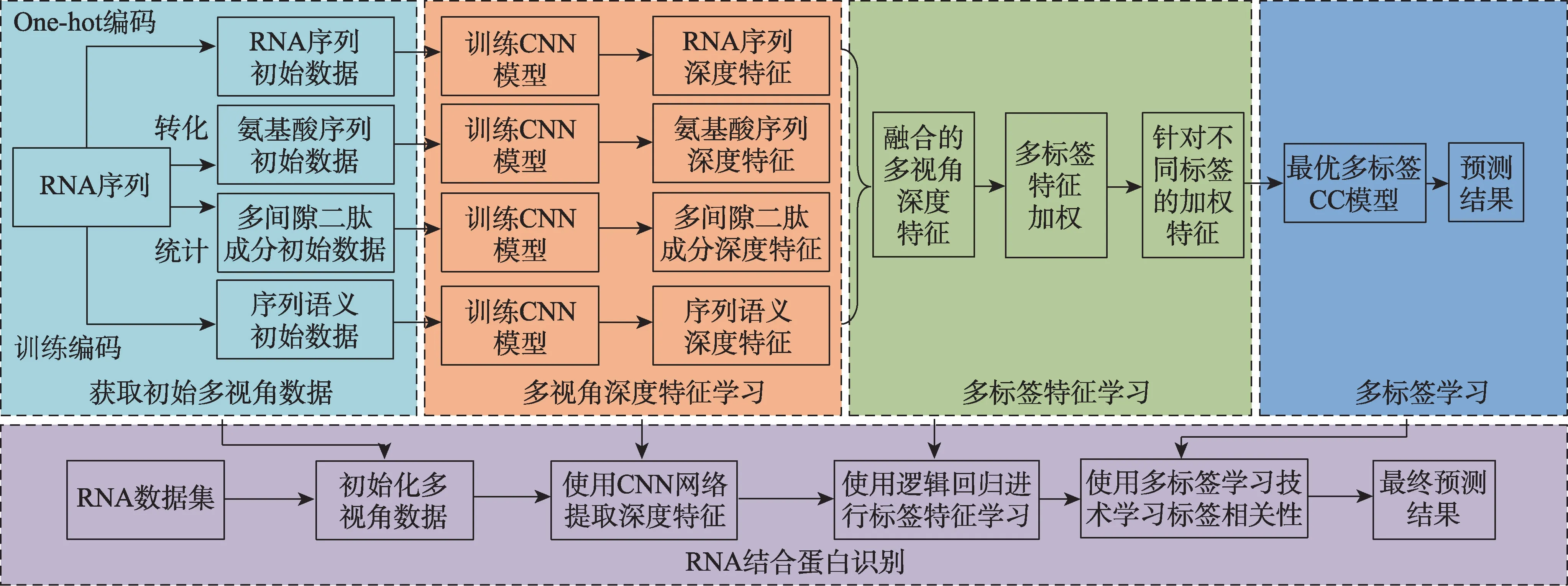
Fig.1 Optimal multi-label chain learning method framework based on multi-view learning图1 基于多视角的最优多标签链式学习方法框架
2.2 多视角初始特征提取
在大数据时代,数据量增多的同时,数据的表示形式也越来越多样化,对样本进行多视角数据提取成为一种趋势。利用多视角数据间相容、互补的性质对其进行更有效的分析成为很多领域的必然需求。本文从4 个角度对RNA 序列进行数据提取工作,分别为包含次序信息的RNA 序列视角、氨基酸序列视角,包含成分信息的多间隙二肽成分视角和包含语义信息的RNA 序列语义视角。
2.2.1 RNA 序列one-hot编码
RNA 序列是由4 种碱基组成的文字序列。许多方法将其作为特征提取的主体,利用one-hot 编码技术将文字序列转化为数值矩阵,再投入至机器学习模型中去训练。One-hot会为一条长度为m的RNA序列构造一个4×m大小的空白矩阵,将每种碱基转化为4维正交基向量,填充至序列的对应位置,如图2所示。
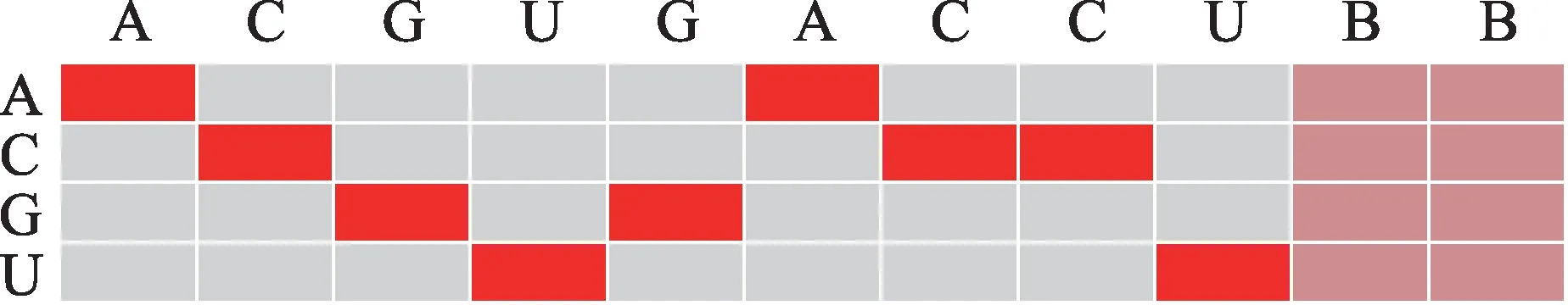
Fig.2 One-hot encoding of RNA sequence图2 RNA 序列one-hot编码
图2 中行标题为一条具体的RNA 序列,实际长度为2 700。对照列中碱基所在的位置,可以把序列中的碱基A 表示为向量(1,0,0,0)T,碱基C 表示为向量(0,1,0,0)T,碱基G 表示为(0,0,1,0)T,碱基U 表示为(0,0,0,1)T,以此类推。由于数据集中RNA 序列的长度不统一,规定了一个固定的长度2 700,使用碱基B 来补齐每条不足2 700 位的RNA 序列,这里统一用向量(0.25,0.25,0.25,0.25)T来表示。
2.2.2 氨基酸序列one-hot编码
上述方法构建的初始矩阵虽然对提取特征有帮助,但缺点是信息量较少。氨基酸序列由20 种氨基酸构成,其信息量远比RNA 序列丰富,因此使用氨基酸序列转化得到的one-hot 编码矩阵会为特征提取提供更好的效果。利用基于codon[29]的三种翻译方式,可将长度为m的RNA 序列转化为3 条长度为1/3m的氨基酸序列,拼接三种形态的氨基酸序列,能够完全继承原始RNA 序列的序列信息,且具有更加丰富的表现形式。对拼接的长链进行one-hot 编码,原理同RNA 序列,可得到20×m大小的初始特征矩阵,如图3 所示,即为本文所提出的氨基酸视角数据。图中行标题为一条具体的氨基酸序列,实际长度为2 700。对照列标题中氨基酸所在的位置,可以将行序列中的所有氨基酸表示为20 维的标准正交基向量,其中i表示氨基酸α在图中列标题所在位置,如氨基酸A 可以表示为氨基酸H 可以表示为氨基酸M可以表示为以此类推。由于RNA 序列存在终止密码子,且部分RNA 序列含有临时碱基B,这里使用字母O 来表示它们,向量值全部设为0.05。
2.2.3 提取序列成分构造多间隙二肽柱状图
上述提到的RNA 视角和氨基酸视角数据偏向于对序列次序提取特征,而序列除了次序外,其组成成分同样重要。因为0-gap 二肽偏向于二维序列的成分组成,而1-gap 二肽带有三维结构成分信息,所以使用0-gap 二肽和1-gap 二肽提取成分信息效果最佳,本文采用它们的组合形式构成多间隙二肽成分视角。二肽对左右氨基酸排列是敏感的,对于本文中21 种氨基酸(20 种天然氨基酸和本文增加的临时氨基酸O),共有21×21×2 个多间隙二肽种类,由于OO 和O*O 的组合对本文的研究无太多意义,被舍弃。统计这880 种二肽出现的次数得到特征向量,可以有效地捕获序列成分信息和空间成分信息。由于880 维的特征向量是一维的,用于提取深度特征的效果不理想,将其转化为二维柱状图,可以更有效地提取深度特征,如图4 所示。图中上部分表格的横坐标为二肽种类,其中“AA”表示左右都是丙氨酸的0-gap二肽,18 代表其在样本序列中的数量;“A*D”表示左侧为丙氨酸,中间间隔任意一个氨基酸,右侧为天冬氨酸的1-gap 二肽。图4 只列举了12 种二肽,实际数量为880 种。下部分图表为转化后的柱状图,每种二肽数量的上限设为30,因此取30×880 大小的矩阵作为此条序列的多间隙二肽初始数据。
2.2.4 使用RNA 词向量构建语义矩阵
自然语言处理(natural language processing,NLP)是计算机科学领域与人工智能领域中的一个重要研究方向,从初始数据的角度来看,生物信息学与NLP的研究数据具有相同的形式[30]。因此,可以使用NLP的方法来解决生物信息学中对文本的编码及初始特征构建。本文使用6 聚体RNA 构建语义词库,6 聚体RNA 为6 个连续碱基组成的结构,词库共由46种6 聚体RNA组成。本文使用现流行的Word2Vec技术构建语义模型,其原理如图5 所示。基于本文所用数据集中的92 102 条RNA 序列,逐条对它们进行以下操作:(1)使用6 位碱基为大小的滑动窗口,获取RNA 序列中6 聚体RNA 的排列顺序;(2)对每个6 聚体RNA 进行编码,即它在4 096种形态中的位置(以“AAAAAA”为1,“UUUUUU”为4 096 的规则);(3)将相邻的2 个6 聚体RNA 分别作为特征X和标签Y,投入至语义模型中训练;(4)从训练完的语义模型中提取4 096种6 聚体RNA 的词向量结果;(5)使用词向量替代RNA 序列中每个6 聚体RNA,构建RNA 序列语义矩阵。由6 聚体RNA 词向量构成的RNA 序列语义矩阵不仅具有较小的维度,而且包含了以6 位碱基为基序的RNA 序列次序和上下文结构信息,可以更好地进行深度特征学习。

Fig.5 Process of generating RNA sequence semantic matrix图5 RNA 序列语义矩阵生成过程
2.3 建立深度卷积网络模型提取深度特征
基于上述4 个视角得到的初始特征数据,构建了4 个不同的深度卷积神经网络模型,来获取这4 个视角的深度特征。深度卷积神经网络(CNN)是一类包含卷积计算且具有深度结构的前馈神经网络,处理对象主要为图像数据,其具有强大的表征学习能力,能够按其阶层结构对输入信息进行平移不变分类[31-33],因此也被称为“平移不变人工神经网络”。4 个视角的模型很相似,输入层之后统一附加卷积层,经过池化、扁平化、dropout 后使用2 个全连通层进行桥接。利用这样的模型所得到的深度特征不仅比原始特征具有更小的维数,而且具有更好的判别能力,增强了后续分类工作的泛化性。图6 中的(1)~(4)分别是提取RNA 序列视角、氨基酸序列视角、多间隙二肽成分视角和RNA 序列语义视角深度特征的CNN 模型图。图中公式k@m×n表示网络各层的特征图数量及大小,k表示该层的特征图数量,m×n表示该特征图的大小;卷积核的大小表示为k×m×n,其中k代表卷积核的数量,m×n代表卷积核的大小,卷积步长默认为1。整个模型的输入为上文提到的各个视角的原始数据矩阵,经过模型处理后得到68 维的特征向量,对应68 个类别(67 种RBP 和不属于任何一类的负类)。CNN 网络最后一层网络采用的激活函数为“Sigmoid”函数,该函数会将最后的全连通层数据映射到0~1 之间,因此最后的特征向量值均为小数,代表RNA 序列隶属于这68 个类的概率分布。网络最后一层只是为了使本文的模型达到拟合数据的目的,深度特征数据经过该层和Sigmoid 函数激活已经有了明显的分类趋势,不利于训练接下来的多标签特征学习模型,因此采用倒数第二层202 维的深度特征作为模型的输出结果。

Fig.6 4 views'deep feature extraction network model图6 4 个视角深度特征提取网络模型
图6模型中,除了最后一层全连通层使用“Sigmoid”激活函数外,其他网络层的激活函数均为“ReLU”。因为“ReLU”函数比“Sigmoid”具有更小的计算量,且有一定的防止梯度消失的作用。由于最后一层要将特征向量与标签相关联,使用“Sigmoid”函数比较合适。“ReLU”函数和“Sigmoid”函数的公式定义如下:

因为多标签分类问题可以看成是由若干个二分类问题所组成,所以使用二分类的损失函数来处理多分类问题。上述4 个CNN 模型均采用二进制交叉熵(binary_crossentropy)作为损失函数,定义如下:
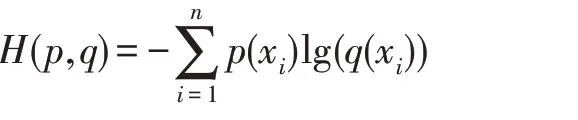
其中,p(xi)和q(xi)都代表序列x对于类别i的隶属度,p代表实标签值,即1 或0,q代表预测值,在这里因为经过“Sigmoid”函数激活,所以q∈[0,1]。
2.4 基于多视角的最优多标签链式学习算法
CC 算法是一种可以高效学习标签之间关联的多标签分类算法,其原理是构建若干个二分类器来预测对应的若干标签,每训练完一个二分类器,算法都会将该分类器预测的对应标签结果附加到初始特征之后,作为下一个二分类器训练的输入特征,直至所有分类器训练完毕。然而CC 算法存在三种弊端:其一,每次为新标签训练分类器时,尽管加入了部分已预测的标签值作为新特征,然而输入的初始特征始终不变,无法最大化利用标签关联性训练新标签分类器;其二,CC 算法过于依赖标签排列顺序,最初的标签分类器性能直接影响了后续新标签分类器的训练效果;其三,现有的CC 算法无法应用于多视角数据场景下的多标签学习。
鉴于此,本文改进了现有CC 链式分类器,将其应用到多视角场景。利用多标签特征学习技术,把多视角数据的优势附加到CC 算法中,同时最大化提升了分类器对每个标签的学习能力,使之可以更好地学习标签之间的关联,达到最优多标签链式学习的效果,具体原理如图7 所示。

Fig.7 Principle of optimal multi-label chain learning based on multi-view learning图7 基于多视角的最优多标签链式学习原理
算法分两部分:多标签特征学习和多标签学习。首先从上游的CNN 模型获取各个视角的深度特征向量,将它们拼接并投入至多标签特征学习网络模型中训练。该模型的输入为808 维特征向量,输出为68维结果,对应68 个标签。通过该模型的学习,可以获取68 组808 维的权重系数,对应输入向量的每一维特征对预测每个标签的贡献权重。将808 维特征向量依次与这68 组权重系数相乘,获得68 组加权特征向量,用于训练下游的CC 多标签分类器。本实验的CC 多标签分类器由68 个二分类器组成,用于预测一条RNA 对68 个标签的隶属情况。鉴于CC 多标签分类器对标签顺序具有依赖性,根据训练集中各标签样本数量的差异对标签分类器的训练进行预排序,使得样本数量较多的标签分类器始终处于优先训练的状态。根据此训练次序,从多标签特征学习模块获得加权特征向量x1,并将其用作输入特征开始训练第一个二分类器。由它预测的第一个标签值被附加到加权特征向量x2末尾,用以训练第二个二分类器。重复该过程,直至最后一个二分类器训练完毕。不同于传统的CC 多标签分类器,本文提出的最优CC 多标签分类器,其特点在于,当训练完第i个二分类器后,将目前预测的所有标签值附加到与下个标签关联的加权特征向量xi+1的末尾,进行第i+1 个二分类器的训练。这样不仅保留了CC 算法学习标签关联性的能力,而且最大化提升了CC 多标签分类器对每个标签的学习能力,把多视角和多标签算法的优势结合在一起,形成最优多标签链式学习。训练和预测算法如算法1、算法2 所示。
算法1 最优CC 多标签分类器训练过程(基于L个标签和与L个标签相关联的加权特征向量数据集DL)


算法2最优CC 多标签分类器预测过程(基于样本X的L组权重特征)

3 实验研究
3.1 数据集
本文使用的数据来源于AURA 网站[34]。本文从该网站上获取了137 003 条RNA 序列信息,1 264 种调控因子信息以及2 549 510 个它们之间的结合位点信息,如图8 所示,红色整圆部分代表RNA 序列库,结合位点信息和调控因子信息属于绑定关系,因此使用蓝色半圆和绿色半圆分别表示它们,图中各颜色数值代表对应部分的信息数量。调控因子又称反式作用因子,是转录模板上游基因编码的一类蛋白调节因子,包括激活因子和阻遏因子等。常见的调控因子有RBP、miRNA、转录因子。因本文是研究RBP 结合关联性问题,且上述结合位点信息中涉及的RNA 不全包含在137 003 条序列中,所以最终本文选取了67 种RBP,73 681 条RNA 序列信息和550 386个它们之间的结合位点信息,如图8 中双圆的交叉部分所示。除此之外,本课题加入了18 421 条没有任何结合位点信息的RNA 序列并入数据集,作为负样本使用。
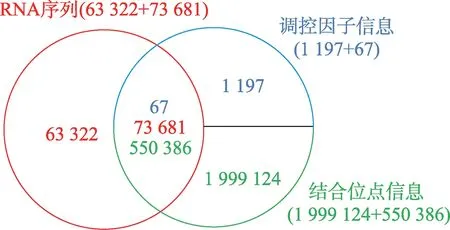
Fig.8 AURA database composition图8 AURA 数据库组成
3.2 性能指标
本文采用AUC 面积和F1 得分两种评价指标来评价模型的分类性能和预测性能。AUC 被定义为ROC 曲线下面积,ROC 曲线是反映敏感性和特异性连续变化的综合指标[35],AUC 值越大,模型的分类性能越好。在多标签分类问题中,类别样本不均衡会导致性能指标偏差过大,因此本文引入常用的Macro、Micro 和Weight 约束条件。Macro-AUC通过给每个类设置相同的权重,计算各个类的AUC 求和均值得到,当小类很重要时该数值会偏低。Micro-AUC 是将每个类的敏感性和特异性分别求和,得出的结果绘制成ROC 曲线,求得的AUC,当大类很重要时该数值会偏低。Weighted-AUC 根据每个类的样本数量,计算出每个类的权重,再对这些类的AUC 进行加权求和。除了评价模型分类性能外,还将对模型的预测性能进行F1-score 的计算,F1 值是准确率和召回率的调和平均数,和AUC 指标相同,也为F1 增加了Macro、Micro 和Weight的约束条件。
3.3 本文算法预测效果
为探究一条未知RNA 与多种RBP 的结合情况,Pan 等人使用多标签技术,利用卷积神经网络和长短时记忆网络建立了iDeepM[11]预测模型,本文在相同数据集的基础上对提出的方法RRMVL 以及RRMVL下各个视角的单视角模型进行五折交叉验证测试。同时,为了验证深度学习在基于长样本RNA 序列上的学习优势,本文构造了编码整条RNA 序列作为特征进行训练的决策树分类模型,以此与深度学习模型进行对比,结果如表1 所示。k折交叉验证在确保一致的数据分布的基础上,将数据划分为大小相同的k个子集。每次将其中一个子集作为测试集,其他子集作为训练集。获取k个测试结果的平均值作为最终结果。这种验证方法有效避免了试验样品的取样偏差,从而获得了更有说服力的试验结果。
从表1 可以看出,使用深度学习的iDeepM 模型和RRMVL 模型效果均优于决策树模型,证明深度学习在提取长样本特征上的优势明显。RRMVL 方法下个任意视角模型预测值均优于iDeepM 模型,体现了本文方法的有效性。同时,所有视角模型整合下的RRMVL 方法在AUC 数值和F1 数值上均比任意单视角模型高,体现了多视角数据之间的信息互补性,同时也说明数据的多视角化在生物信息学领域可以取得较好的效果。从单视角来看,多间隙二肽成分视角取得了最好的效果,这是因为多间隙二肽不仅包含序列次序信息,而且包含了序列成分和结构信息,是信息量最丰富的视角。RNA 序列语义单视角的效果相比初始RNA 序列视角略低,这是由于通常训练一个好的语义模型需要百万级的样本数据,而本文的数据集仅包含92 102 条RNA 序列,不足以训练出效果理想的6 聚体RNA 词向量,因此预测性能效果不佳。总体而言,在3 种对比算法中,本文提出的RRMVL 方法取得了3 项AUC 和3 项F1 的最佳效果,由此证明基于多视角的最优多标签链式学习方法在识别RNA 结合蛋白的问题上达到了预期效果。
3.4 多标签特征学习和多标签学习有效性分析
为检验本文使用的多标签特征学习和最优多标签链式学习效果,本文在AURA 数据集上对RRMVL及其变体方法进行了双重对比实验,分别为使用基于多视角投票的集成学习RRMVL 方法与使用多标签特征学习RRMVL 方法对比,以及未使用多标签学习的RRMVL 方法和使用多标签学习的RRMVL 方法对比。因基于多视角投票的集成学习模型不是一种分类器,所以没有AUC 指标,其余方法的五折交叉验证结果如表2 所示。

Table 1 Performance of algorithms on AURA dataset表1 各算法在AURA 数据集上的性能

Table 2 Effect comparison of multi-label feature learning and multi-label learning表2 多标签特征学习、多标签学习效果对比

Fig.9 Line charts of methods'performance comparison for single class dataset图9 单个类数据集的方法性能对比折线图
从表2 可以看出,对于多视角数据而言,在对其使用多标签特征学习后,模型的预测性能始终比基于投票的集成学习突出,说明多标签特征学习充分利用了多视角数据的优势。另一方面,在处理多标签分类问题上,使用多标签分类器的方法始终优于未使用多标签技术的方法,证明了标签之间的关联对预测产生了不可忽视的作用。结合多标签特征学习和多标签学习,即本文提出的最优多标签链式学习,其性能优于使用集成学习的传统CC 算法,证明本文对传统CC 算法的改进富有成效。值得注意的是进行多标签学习后,RRMVL 的AUC 指标有所下降,这是由于多标签CC 分类器的分类性能与神经网络最后一层的“Sigmoid”网络分类能力略有差距。对于三项F1 指标,基于最优多标签链式学习方法RRMVL取得了最好效果,再次证明本文方法能够较为准确地判别某条未探索的RNA 与多种RBP 的结合情况。
3.5 类样本不均衡影响分析
为研究类样本数量对实验效果的影响,本文使用RRMVL 对68 个类数据集进行单独实验,对比iDeepM 的五折交叉验证结果折线图如图9所示。图9为准确度、召回率和F1 指数的折线图,按照类样本数量递增的次序进行绘图。
从图9 可以看出,两种对比算法中,RRMVL 在大部分类的预测精度取得了最佳效果,两个方法随着类样本数量的逐渐提升,各指标都呈现逐渐提高并趋于平缓的趋势。当样本数量低于5 000 时,各项指标的起伏较大,这是由于类样本数量过少导致模型无法准确地学习这些类样本的深度特征。对比两条曲线,iDeepM 方法在低样本环境下的学习能力不如RRMVL,表现为动荡幅度更剧烈,间接体现多视角数据在小样本学习下的优势。总体而言,本文方法在各个类数据集上达到了预期效果。
4 结论和展望
本文提出一种基于多视角的最优多标签链式学习法来对一条未探索的RNA 进行RBP 结合性识别,通过实验数据可以发现,本研究提出的多视角深度特征提取法对比传统的特征提取,获得了更好的效果。并且使用多标签特征学习和最优多标签链式学习的算法进一步提高了预测精度。可以发现以多间隙二肽成分视角为首的多视角数据以及多视角多标签的学习方法对识别RBP 具有较大的价值。
虽然研究的效果得到了提升,但是该研究还有一些不足和值得进一步深入研究的地方。比如RNA序列语义性视角的性能略低,没有达到预期的效果。随着测序工作的进行,未来基于大数据下的语义模型能够更好地学习到6 聚体RNA 的词向量分布,该视角的预测精度可以得到一定的提升。此外,本文所用数据集中不同类别的样本数量相差过大,属于典型的类不平衡问题,对模型的学习效果和分类效果产生较大影响。未来如何构造更适合类不平衡场景下的RBP 识别方法也将是一个重要的研究方向。
