中孕期母血产前筛查的室内质控结果室间比对分析
2021-11-13段红蕾李照侠朱雨捷
张 颖,段红蕾,李照侠,朱雨捷,李 洁
南京大学医学院附属鼓楼医院产前诊断中心,江苏南京 210008
中孕期母血清学筛查主要通过检测母血清中甲胎蛋白(AFP)、游离人绒毛膜促性腺激素(Freeβ-hCG)水平,同时结合孕妇年龄、体质量、采血孕周等妊娠相关信息,评估胎儿21-三体、18-三体、开放性神经管畸形的患病风险[1-2]。实验室对AFP、Freeβ-hCG指标检测的准确性,将直接影响筛查结论及后续的临床处理,严格的室内质量控制(简称质控)管理和室间质评是高质量产前筛查工作中的重要环节[3]。为更好评估实验室检测的精密度和准确度,在已有室内质控和室间质评的基础上,江苏省在全省范围内开展孕中期母血清学筛查数据室内质控室间比对工作。
1 资料与方法
1.1数据采集 在江苏省产前筛查质控数据平台上,收集2018年9月至2019年8月33家检测单位上报的中孕期母血清学产前筛查的AFP、Freeβ-hCG质控数据9 632个。
1.2质控品 各实验室统一使用相同批号的AFP/Freeβ-hCG质控(U2180501,杭州宝荣)。每套质控品包括高、中、低3个水平。
1.3检测方法 各家实验室将统一批号的质控品和标本同时采用时间分辨免疫荧光法进行检测。各检测单位按照Levey-Jennings质控标准根据检测结果完成室内质控后,将质控品测定数据及仪器型号上传至江苏省产前筛查质控数据分析平台。
1.4试剂与仪器 检测仪器及试剂均由美国PerkinElmer公司生产。33家检测单位中,21家单位(A1~A21)使用1235型仪器,4家单位(S1~S4)使用1420型仪器,8家单位(D1~D8)使用DX型仪器。
1.5评价指标 根据国家标准《临床实验室室间质评要求》制订评定标准[4]。
1.5.1精密度评价 变异系数(CV)=标准差/均值。变异系数指数(CVI)=某个实验室标准差/所属仪器分组标准差。判定标准:CV<5.0% 且CVI≤1.0,为合格;CV<5.0%且CVI>1.0,为假阳性;CV>5.0%且CVI>1.0为真阳性。CV的合格需结合CVI值进行综合分析。
1.5.2结果一致性评价 总误差(TE)=|单个实验室均值-所属仪器分组均值|+2×单个实验室标准差。允许总误差(TEa)设定为实验室所属仪器分组均值的30%。
标准差指数(SDI)=(单个实验室均值-所属仪器分组均值)/所属仪器分组标准差。判定标准:TE≤TEa且|SDI|≤2.0为合格,TE≤TEa且|SDI|≤1.0为良好,TE≤TEa但|SDI|>2.0或TE>TEa均为不合格。
1.5.3准确度和精密度综合评价 临界系统误差(ΔSEc)= (所属仪器分组TEa-|单个实验室均值-所属仪器分组均值|)/单个实验室标准差-1.65,ΔSEc用于准确度和精密度的综合性评判,测量在5.0%以上结果超出TEa界限之前均值偏移的标准差个数。 判定标准:ΔSEc>2.0为合格。如ΔSEc≤2.0,需重新制订实验室的质控规则。
1.6统计学处理 采用R软件(4.0.2版本),统计AFP和Freeβ-hCG低值、中值及高值质控品的测定个数、均值、标准差、CV、SDI、CVI、TE和ΔSEc等参数,并采用ggplot2包分别绘制AFP和Freeβ-hCG指标的CV值分布直方图,以及CVI和SDI的尤敦图。
1.6.1纳入标准 因各实验室标本量不同导致实验次数不相同,纳入同一质控品批号使用频次≥10次的数据。
1.6.2剔除标准 根据Grubbs检验法的临界值表剔除极端异常值:G=|单个实验室测定值-单个实验室均值|/单个实验室标准差。剔除超过本组均值±3标准差的质控数据。
2 结 果
2.133家实验室的精密度测定分析 AFP检测的精密度测定合格率为97.0%(32/33,图1),不合格率为3.0%;Freeβ-hCG检测的精密度合格率78.8% (26/33),不合格率为21.2%。其中编号S1的实验室中AFP和Freeβ-hCG的3个水平精密度均不合格,均CV>5%。Freeβ-hCG测定中发现,共7家实验室Freeβ-hCG质控结果不合格,其中S1实验室3个水平质控结果均不合格,D7、D8、A4实验室分别有2个水平质控结果不合格,D2、D6、A13实验室各有1个水平质控数据不合格, 且CV均>5%(图2)。
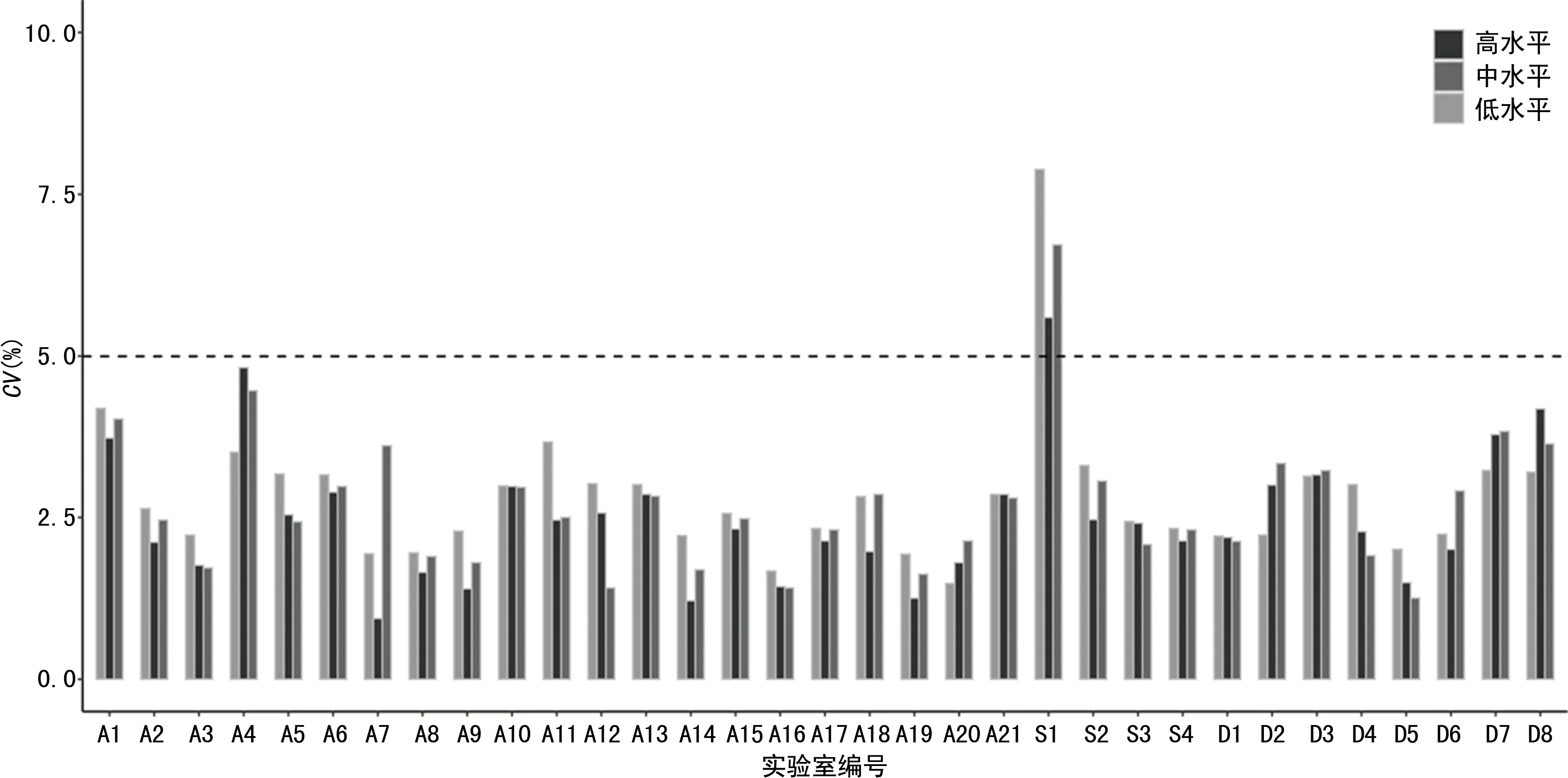
图1 33家实验室AFP检测的CV分布图
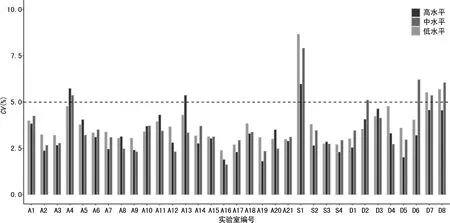
图2 33家实验室Freeβ-hCG检测的CV分布图
2.233家实验室测定结果的一致性分析 33家实验室AFP和Freeβ-hCG检测均为TE≤TEa且|SDI|≤2.0,AFP和Freeβ-hCG检测一致性合格率为100.0%。其中23家实验室AFP测定TE≤TEa且|SDI|≤1.0,实验室一致性测定良好率为69.7%(23/33,图3);Freeβ-hCG检测的实验室一致性测定良好率为81.8%(27/33,图4)。
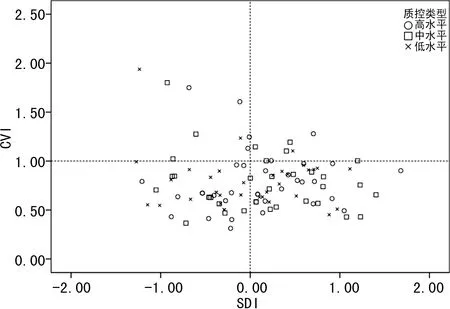
图3 33 家实验室AFP检测的CVI与SDI分布尤敦图
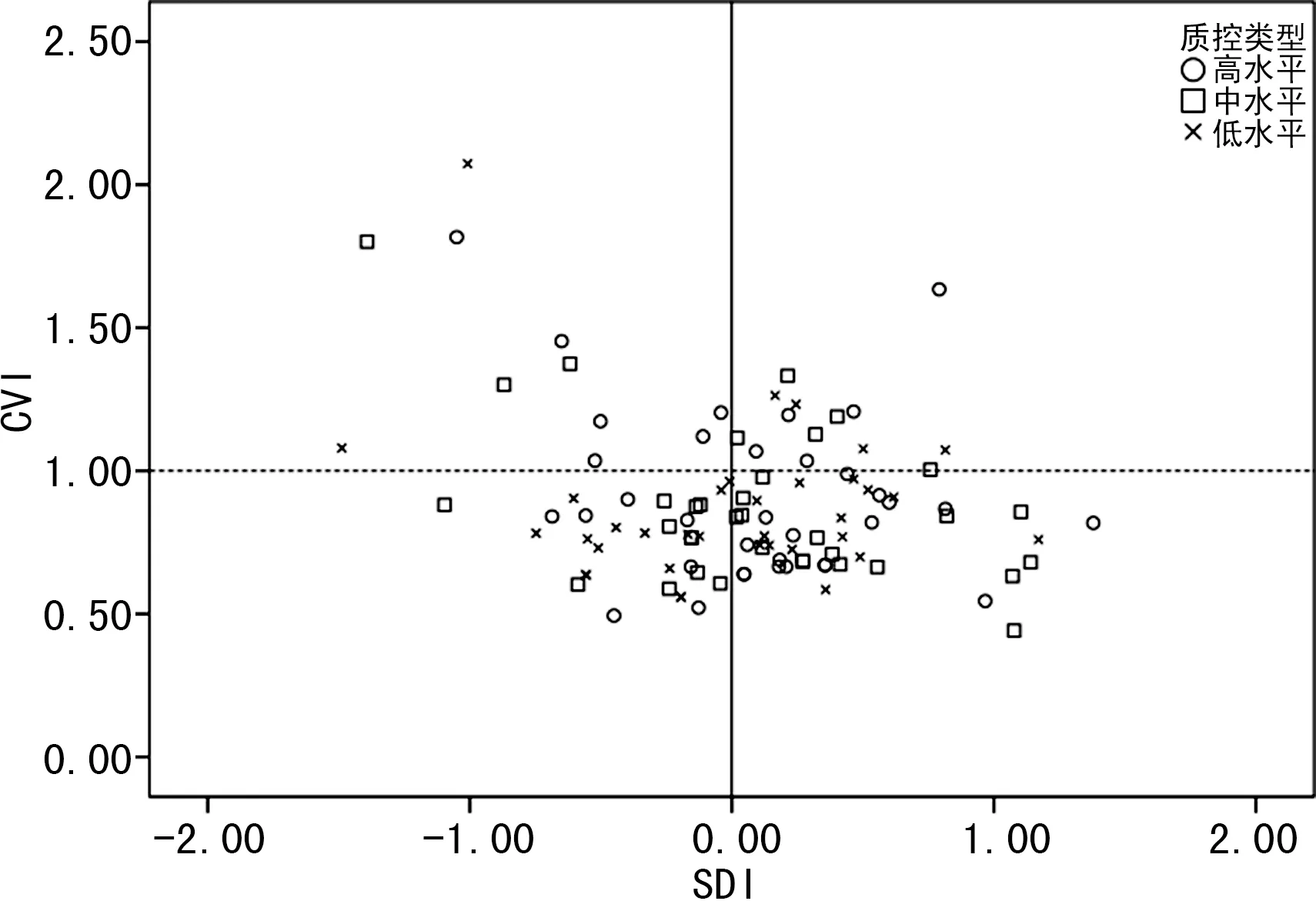
图4 33 家实验室Freeβ-hCG检测的CVI与SDI分布尤敦图
2.33种仪器测定的精密度分布 剔除138个数据,纳入9 494个质控数据。1420型仪器组质控剔除率(3.19%)高于1235型仪器组(1.24%)和DX型仪器组(1.65%),差异有统计学意义(χ2=26.20,P<0.001);3组仪器间,AFP的CVI合格率差异无统计学意义(χ2=2.36,P=0.308);Free β-hCG的CVI合格率比较:DX型仪器组低于1235型和1420型仪器组,差异有统计学意义(χ2=6.94,P=0.031)。
各仪器组的SDI和TE都符合基本要求。1235型仪器组和DX型仪器组ΔSEc达标率100%(29/29);1420型仪器组ΔSEc有1家实验室(S1)不达标。见表1。

表1 3种仪器测定AFP和Freeβ-hCG的精密度分布

续表1 3种仪器测定AFP和Freeβ-hCG的精密度分布
3 讨 论
质量管理是实验室管理的核心,搞好质量管理也是实现实验室标准化、规范化和科学化管理的关键。实验室可采用室内质控来评价检测结果的精密度,但较难评价准确度;采用室间质评可进一步评估检测的准确度,但频次相对较低。本次是对2018年9月至2019年8月江苏省33家产前筛查实验室使用相同批号的质控品,并将室内质控结果实时上传做室间质量比对,可高频率同时监测评价实验室检测的精密度和准确度,较好地弥补了室内质控和室间质评的局限性[5]。
CV是常用的精密度评价指标,本研究中AFP和Freeβ-hCG指标CV不合格率分别为3.0%和21.2%。CV不达标,说明实验室检测稳定性差,随机误差大。实验室环境、仪器状态、加样量是否准确等因素均会影响检测的精密度。CVI被用来评价相同仪器组内各实验室检测的相对精密度。CVI的假阳性或假阴性评估,需要与该组的组CV来比对:组CV比较小,那么可能出现单家实验室CVI假阳性,反之组CV很大,则可能出现单家实验室CVI假阴性。使用1235型仪器的实验室中有7家(A1、A4、A5、A7、A10、A11、A13)、使用DX型仪器的实验室中有5家(D2、D3、D4、D7、D8)都出现CVI假阳性,这是其组内均值较低造成的。因此在分析单家实验室CV时需结合组内CVI。Freeβ-hCG指标检测CV值较大,可能与Freeβ-hCG指标更易受温度等因素影响有关[6]。有研究表明,温度升高可导致Freeβ-hCG假性增高,进而导致21-三体假阳性率增加;反之增加漏诊风险。
TE是总误差,含精密度和准确度;SDI被用来评价相同仪器组内各实验室检测的相对准确度。A11、A12、A16、S1实验室SDI虽达标,但检测值相对本组均值偏离较大,需警惕。以A11实验室为例,AFP指标偏低会引起21-三体和18-三体阳性率升高,导致开放性神经管畸形阳性率降低。A12实验室AFP和Freeβ-hCG指标均偏高,导致增加18-三体漏诊风险。建议上述实验室通过对仪器的加样针进行校准,调整标本处理流程等方式降低系统误差,同时评价所在地区人群分布中位数来决定是否需要调整中位数方程[7],降低与同组实验室的差异。
除S1实验室外,本次各家实验室ΔSEc均达标,说明使用的质控规则和方法以及设定测定的质控品个数符合质量控制规范标准。
仪器分组结果显示,在不考虑S1实验室的情况下,3种技术平台均具有较好的一致性。在精密度方面,1235型仪器组优于1420型仪器组和DX型仪器组。1420型时间分辨荧光分析仪属于半自动化机器,需要手工加样,孵育的时间和温度误差可能容易对检测稳定性产生影响。DX型与1235型虽然都是全自动仪器,1235型每批次实验均进行定标,而DX型用户大多采用定期定标校准方式,如果仪器状态改变却未及时定标校准,可能对检测结果造成影响,所以实验室应定期对仪器做维护,及时发现问题;同时DX型投入临床使用时间相对于1235型较短,数据量较少,目前使用定期定标的方法是否需要调整,有待于进一步积累数据。
综上所述,本次实验的室内质控结果室间比对显示,除S1实验室外,其他实验的精密度检测均合格,同一检测平台的检验结果也具有较好的一致性,但不同实验室、不同检测平台的精密度尚存在一定差异,可通过SOP固化流程、人员培训熟练操作、仪器维护与保养、设备更新和性能增高等措施来改进。该方法可以发现无室间质评检测项目在同一检测平台的报告结果中存在的质量问题、检验结果的不一致性,有利于提高检验结果的可比性。
