靶向肿瘤驱动抗原的T细胞受体工程化T细胞治疗策略
2021-11-13罗凌杰汤宇翔张周朝阳梁文华谭润琳钱威瑾王锋
罗凌杰,汤宇翔,张周朝阳,梁文华,谭润琳,钱威瑾,王锋
(上海交通大学医学院 上海市免疫学研究所,上海 200025)
近年来,以程序性死亡受体1(programmed death-1,PD-1)等免疫检查点阻断抗体和嵌合抗原受体T细胞(chimeric antigen receptor T cells,CAR-T)免疫疗法为代表的肿瘤免疫疗法已在临床上取得了重要进展[1-2]。免疫检查点阻断抗体疗法以T细胞的免疫检查点PD-1/程序性死亡配体1(programmed death-ligand 1,PD-L1)及细胞毒性T淋巴细胞相关蛋白4(cytotoxic T lymphocyte associate protein 4,CTLA-4)为靶点,诱导T细胞增强抗肿瘤免疫应答[3]。免疫检查点阻断抗体疗法常伴随有自身免疫性疾病的发生,成为临床治疗中的一个难点,而以基因工程技术为基础的T细胞治疗可使这一副作用发生率大大降低。在CAR-T疗法中,通过基因工程技术,将1个T细胞激活域[通常包括分化簇-3(cluster of differentiation-3,CD3)复合物的ζ链]和细胞外免疫球蛋白衍生的重链和轻链组合形成嵌合抗原受体(chimeric antigen receptor,CAR),并通过将其移植于T细胞使T细胞获得不受主要组织相容性复合物(major histocompatibility complex,MHC)限制的抗原识别能力,并通过这一识别,活化T细胞,从而发挥其抗肿瘤的功能。但由于第1代CAR-T活化能力受限,无法长时间发生T细胞反应,所以在第1代CAR-T的基础上,通过添加共刺激分子胞内域及细胞因子信号域,发展出了拥有1个共刺激分子的第2代CAR-T、拥有2个甚至更多共刺激分子的第3代CAR-T及具有细胞因子或趋化因子受体的第4代CAR-T,大大增强了T细胞的活化和存活[4]。CAR-T识别抗原不受MHC的限制,并只能识别细胞表面抗原,且需要多个靶点的识别才能发挥其抗肿瘤功能,这可能是细胞因子释放综合征这一较为严重的副作用的由来。目前,CAR-T疗法在血液肿瘤的治疗中表现出了巨大的优势,但在实体瘤的治疗中效果尚不明确。同时,免疫检查点阻断抗体疗法在实体瘤中的有效率也亟需进一步提升。而T细胞受体工程化T细胞(T cell receptor-engineered T cells,TCR-T)免疫疗法识别的是多肽-MHC复合体,可精确定位并杀伤呈递这些多肽的细胞,且可靶向所有被MHC呈递的抗原,而不限于细胞表面抗原,尤其是胞内抗原,如肿瘤突变抗原和来源于致癌病毒的抗原。此外,由于TCR-T可识别多肽-MHC复合体,相较于CAR-T疗法,在回输入患者体内时,不会被循环中的抗原阻断,大大提高了TCR-T实体肿瘤归巢的可能。这些特点均可提升TCR-T的抗实体肿瘤功能。本文重点介绍以肿瘤驱动突变、T细胞受体与抗肿瘤免疫应答为基础的TCR-T研究进展,同时介绍驱动突变抗原-TCR验证筛选系统,为更深入的实体瘤TCR-T临床治疗研究提供新思路。
1 肿瘤驱动突变生物学特征
肿瘤细胞基因组中存在多种突变,只有少数能诱导肿瘤发生、发展的突变被定义为驱动突变,这些驱动突变是肿瘤细胞的特征性标志之一,确定肿瘤细胞的驱动突变是肿瘤治疗的关键环节。肿瘤细胞的驱动突变可发生在原癌基因及抑癌基因中。在原癌基因中发生的驱动突变进一步增强了原癌基因的功能,促进了肿瘤细胞增殖,而驱动突变在抑癌基因中解除了其对肿瘤细胞生长调控的抑制作用,诱导肿瘤发生、发展。在原癌基因BRAF中最常发生的V600E突变是包括甲状腺癌、黑色素瘤、结肠癌等多种恶性肿瘤发生的重要诱因[5]。错义突变导致的抑癌基因TP53功能失活也是诱导肿瘤发生的主要因素[6]。此外,对转化生长因子-β(transforming growth factor-β,TGF-β)的研究证实,在肿瘤发生、发展的早期,其信号失活的突变诱导了早期肿瘤的发生,而在肿瘤发生的后期,增强TGF-β信号的突变驱动了肿瘤的进展[7-8]。因此,特定的突变可能只在肿瘤发生、发展的某些阶段起驱动作用,而同一基因向不同方向的突变均可能驱动了肿瘤的形成。
通过对癌症相关突变基因的研究发现,约有90%的癌基因突变发生于体细胞中,此外有20%的癌基因突变发生于生殖细胞中,而10%的癌基因突变可同时在体细胞和生殖细胞中发生[9]。这些存在于生殖细胞中的癌基因突变表明,驱动突变可遗传给子代,生殖细胞发育来的机体在生命早期携带有驱动突变,但绝大部分肿瘤并没有发生在生命早期,即在细胞癌变前数十年这些细胞既已携带有驱动突变,但并没有发生癌变。对多种癌组织样本的基因组数据研究表明,大多数样本携带不止1个突变,其中有一些突变在不同的癌组织样本中高频出现,且在其所研究的30种肿瘤中有25种肿瘤存在与年龄因素相关的标志性基因突变[10]。因此,肿瘤细胞可能并不是由单一驱动突变诱导,需要多种驱动突变同时存在以诱导肿瘤的发生、发展,而这些驱动突变随时间的推移在细胞内逐渐累积,最终导致肿瘤的发生。通过对肿瘤全基因组和肿瘤基因组图谱的分析发现,绝大部分肿瘤发展需要平均4 ~ 5个驱动突变,但在约5%的病例中未检测到已知的驱动突变,这意味着可能还有更多潜在的驱动突变有待发现;在肢端黑色素瘤中,染色体突变性断裂重组先于大多数体细胞点突变,并同时影响多个肿瘤相关基因,这表明肿瘤的形成很可能是多个驱动突变协同作用的结果,单一的驱动突变可能不足以诱导肿瘤生成,通过对肿瘤细胞基因组更为深入的探索,可能发现不同驱动突变在肿瘤发生、发展中的协同特性[11]。
2 T细胞受体肿瘤新抗原的识别对抗肿瘤免疫的作用
在抗肿瘤免疫反应中,T细胞是主要的效应细胞,具有特异性识别肿瘤抗原的能力。T祖细胞(pro-T)从胚肝或骨髓中迁出,并在胸腺微环境中通过T细胞受体(T cell receptor,TCR)识别抗原信号进行胸腺选择,发育为成熟T细胞后随血液循环最终定居于外周淋巴系统或组织器官中。经过胸腺选择后,TCR即获得了对MHC分子呈递的外源或突变抗原识别的能力,又可以排除对自身抗原的识别,从而获得免疫耐受能力。由此形成的每一T细胞克隆均携带有独特的TCR序列,可特异性地识别相应的抗原多肽,数量众多的T细胞表达不同的TCR,从而形成具有丰富多样性的TCR组库。TCR组库的多样性是其抗原特异免疫应答的分子基础,决定了T细胞抗肿瘤免疫应答效应。
2.1 T细胞与T细胞受体信号
TCR是由多个跨膜蛋白组成的蛋白质复合体,其包含了由TCRα与TCRβ链或TCRγ与TCRδ链组成的二聚体以及由4种CD3蛋白组成的3个二聚体(分别为CD3εδ,CD3εγ和CD3ζζ)。外周血中具有TCRα链及TCRβ链组成的T细胞(后文简称:TCRαβ二聚体或αβ T细胞)占总T细胞的90%以上,而具有TCRγ链及TCRδ链TCR组成的T细胞(后文简称:TCRγδ二聚体或γδ T细胞)仅占不到10%[12]。与γδ T细胞不同的是,αβ T细胞与抗原的识别受MHC的限制,在适应性免疫应答中发挥重要作用。αβ T细胞的TCR二聚体主要以胞外区为主,负责识别其他细胞表面的肽段-主要组织相容性复合体(peptide-major histocompatibility complex,pMHC)。因此,TCRαβ-CD3复合体可通过TCR胞外区与抗原-MHC结合,又可通过CD3二聚体将信号传递到细胞内。
TCRα链 和TCRγ链由多个不同的可变区(variable region,V)、铰链区(joining region,J)和恒定区(constant region,C)片段组成,TCRβ链和TCRδ链则由不同的V区、多变区(diversity region,D)、J区和C区片段组成,且每个片段中都有多个不同的基因片段,这些不同的基因片段在重排的过程中随机组合,这是TCR多样性产生的主要机制。TCR与抗原的选择性结合有赖于TCR高特异性的V区,V区有3个超变区,亦称为互补决定区(complementarity determining region,CDR),其中CDR3的变异决定了TCR的抗原特异性,结合TCR的V(D)J基因的重排,使得个体产生庞大的TCR组库。在TCRαβ二聚体对抗原-MHC复合体的识别中,CDR1和CDR2识别MHC分子抗原结合槽中由α螺旋组成的侧壁,CDR3则直接识别MHC所呈递的抗原。
当细胞外的TCRαβ-CD3复合物结合到能与之相互匹配的pMHC,通过胞内的CD3复合物形成抗原特异性信号即第一信号传入,在蛋白酪氨酸激酶(protein tyrosine kinase,PTK)的作用下,TCR自身的酪氨酸磷酸化,由此激活下游信号通路。激活的抗原信号再整合共刺激/共抑制受体产生的第二信号以及细胞因子信号,决定T淋巴细胞的抗原特异性免疫反应或免疫耐受[13]。
2.2 T细胞抗肿瘤免疫应答
在T细胞的抗肿瘤免疫应答中,CD4+T细胞通过CD40L与树突状细胞(dendritic cell,DC)的表面蛋白CD40相结合,诱导DC产生如白介素-12(interleukin-12,IL-12)和白介素-15(interleukin-15,IL-15)的细胞因子,作用于细胞毒性CD8+T细胞,增强其杀细胞功能,直接杀伤肿瘤细胞。T细胞抗肿瘤免疫治疗的关键是靶向抗原肽,抗原肽则是由癌细胞上MHC分子进行呈递。在肿瘤的发生、发展中,肿瘤细胞由突变诱导生成许多抗原肽,在其释放后被DC捕获;DC对这些捕获的抗原进行加工处理,并经MHC分子将抗原呈递给T细胞,通过这些肿瘤来源的抗原活化T细胞;其中活化后形成的效应性CD8+T细胞经循环进入肿瘤组织,通过对MHC-I类分子呈递的同源抗原的识别,杀伤表达这些同源抗原的肿瘤细胞;被杀伤的肿瘤细胞释放更多的肿瘤抗原,以增强这一T细胞抗肿瘤反应[14]。
针对多肽与不同的人类白细胞抗原(human leukocyte antigen,HLA)亚型结合效率及结合位点的差异,TCR对抗原多肽的识别可产生不同的应答效果。只有当HLA亚型有效结合多肽,且被相匹配的TCR识别后,T淋巴细胞才能被激活,从而开启对肿瘤细胞的杀伤。有研究显示,当多肽-MHC与TCR的亲和力较低时,PD-1表达减少,分泌γ干扰素(interferon-γ,IFN-γ)以及肿瘤坏死因子(tumor necrosis factor,TNF)-α的CD8+T细 胞 增 加,从而影响CD8+T细胞对肿瘤的杀伤功能[15];另外,MHC与TCR的持续结合使得CD8+T细胞的共刺激以及共抑制受体在经历短暂的初始上调后,其表达水平便开始下降,从而影响了CD8+T的潜在功能[16],这表明MHC的功能不仅仅只是向T细胞呈递抗原,其与TCR的结合更参与了对CD8+T细胞功能的调节。然而,肿瘤细胞可通过多种机制改变多肽-MHC复合体的表达,以规避MHC与T细胞的结合,逃避T细胞介导的杀伤。抗原多肽在肿瘤细胞内通过抗原加工相关转运体,进而被运输到内质网,MHC-I类分子可选择性结合不同序列的多肽,形成多肽-MHC复合体。在肿瘤组织中抗原加工和呈递机制相关基因和蛋白异常较为常见,可导致一些肿瘤相关抗原不能被运输到内质网内并被MHC-I类分子结合,这大大减少了肿瘤抗原-MHC复合体在肿瘤细胞表面的表达[17]。此外,对乳腺癌及非小细胞肺癌的研究表明,肿瘤细胞可下调甚至完全丧失MHC-I类分子的表达,使其逃避了肿瘤抗原特异性CD8+T细胞的识别和杀伤[17-18]。
3 T细胞靶向肿瘤突变抗原
基于二代测序技术的全外显子测序和大panel测序可获知肿瘤样本中每百万编码DNA中体细胞基因突变的总数,即肿瘤突变负荷(tumor mutation burden,TMB),它包括同义突变和非同义突变[19]。较高的TMB值提示肿瘤细胞产生较多肿瘤抗原的可能及更多的能被T细胞识别的肿瘤抗原。通过对来源于不同肿瘤类型的多个肿瘤组织样本的分析发现,在突变频率最为显著的黑色素瘤中,每百万编码DNA中有超过10个非同义突变,等同于肿瘤细胞基因编码区内有150个非同义突变[20]。MC38肿瘤模型的研究表明,只有10%的非同义突变产生的多肽可与MHC-I类分子高亲和力结合,而这10%中也只有很少的一部分能诱导免疫应答[21]。目前,所检测的绝大部分肿瘤的TMB小于10[20]。因此,肿瘤细胞产生的能诱导免疫应答的肿瘤抗原极少。
在早期T细胞抗肿瘤研究中,对于目标抗原的选择集中在一些肿瘤中高频出现的肿瘤抗原中。在难治性转移性乳腺癌的治疗中,通过对靶向SLC3A2,KIAA0368,CADPS2和CTSB的 突变蛋白肿瘤浸润T细胞的研究发现,这些T细胞可以对突变产生反应并有效杀伤肿瘤,且大部分情况下这些T细胞可以在外周血中持续较长时间[22]。
4 T细胞靶向致癌病毒抗原
爱泼斯坦-巴尔病毒(Epstein-Barr virus,EBV)、高危的人乳头瘤病毒(human papilloma virus,HPV)亚型、默克尔细胞多瘤病毒(Merkel cell polyomavirus,MCPyV)、乙型肝炎病毒(hepatitis B virus,HBV)、丙型肝炎病毒(hepatitis C virus,HCV)、卡波西肉瘤相关疱疹病毒(Kaposi,s sarcomaassociated herpesvirus,KSHV)及人类T淋巴细胞白血病病毒1型(human T-lymphotropic virus type-1,HTLV-1)的感染是部分肿瘤患者的主要致病因素,这些病毒被认为是致癌病毒。其中,HPV癌基因蛋白E6和E7特异性地表达于宫颈癌细胞[23];其通过p53和pRb的降解,E6和E7与p53和pRb结合的减弱及端粒酶活性的上调,进而诱导角质形成细胞的癌变,从而导致HPV相关恶性肿 瘤 的 发 生[24-27]。EBV核 抗 原1(Epstein-Barr nuclear antigen 1,EBNA1)及潜伏膜蛋白(latent membrane protein,LMP)1和LMP2被 认 为 是导致B细胞恶性肿瘤发生的关键致癌因素[28]。MCPyV阳性的默克尔细胞癌中,肿瘤细胞常表达大T抗原突变体[29-30]。研究表明,MCPyV大T抗原的这一突变体和MCPyV的小T抗原蛋白均显示了强大的致癌能力[31-32];它们是MCPyV阳性的默克尔细胞癌肿瘤细胞存活的必要前提,大T抗原突变体和小T抗原的敲除可诱导MCPyV阳性的默克尔肿瘤细胞的死亡[33-34]。在肝癌的发生、发展中,鉴于HBV和HCV感染导致的肝脏慢性炎症,其编码的蛋白可能不是通过直接诱导而是间接调控的方式介导了肿瘤的发生[35]。这些证据表明,致癌病毒抗原是导致肿瘤发生的主要因素,同时这些抗原只存在于肿瘤组织中,不存在于正常细胞,并且由于病毒基因组编码蛋白序列与人类蛋白序列相似度极低,受淋巴细胞的中枢耐受影响较小,因此致癌病毒抗原具有很强的免疫抗原性,是TCR-T疗法的理想靶点。
5 T细胞靶向肿瘤驱动突变抗原
驱动突变是肿瘤发生、发展的决定性突变,在肿瘤细胞中驱动突变诱导细胞产生带有特异性突变的氨基酸序列,这些突变所致抗原通常是肿瘤所特有的,只表达于肿瘤组织,因此这些氨基酸序列构成的多肽对于免疫系统是完全陌生的,且不在胸腺表达,不会在胸腺T细胞阴性选择过程中被清除。这保证了免疫系统对其具有应答能力,且不会诱导T细胞在中枢免疫过程中对其产生耐受,因此可以产生更为有效的抗肿瘤T细胞免疫应答。此外,由于驱动突变抗原的肿瘤限制性表达,由其引起的免疫应答作用于肿瘤组织,不影响正常的自身组织,可大大降低由其引起自身免疫反应的危险性[36]。
在黑色素瘤的过继T细胞治疗中,肿瘤细胞通过减少抗原表达或等位基因突变,导致原T细胞识别的肿瘤抗原在细胞中选择性地丢失,表明T细胞在肿瘤的发生、发展中对肿瘤抗原的产生和编辑施加了选择压力[37]。这一选择压力在肿瘤细胞的进一步变异中干预了肿瘤抗原的突变,使其逃避了T细胞的杀伤。在TCR肿瘤抗原识别中,以乘客突变(passenger mutation)所致肿瘤抗原为靶标时,随着其突变的持续发生可造成T细胞识别的脱靶。鉴于驱动突变是成为肿瘤的决定性因素,因此以驱动突变抗原为靶标时,TCR识别脱靶的概率远低于乘客突变,这也使得驱动突变成为较为稳定的T细胞识别靶标,有效降低了肿瘤治疗中免疫逃逸的概率。
肿瘤细胞突变所产生的多肽在胞内被蛋白酶体处理后可与MHC结合,这些突变所产生的多肽均可呈递给T细胞。在肿瘤细胞中,乘客突变等非决定性的突变所产生的多肽占肿瘤细胞表面MHC呈递抗原的绝大多数,虽然驱动突变多肽占比较少,但一旦其与MHC结合表达于细胞表面,即可被T细胞识别,这是由TCR的特异性决定的。通过对P53的驱动突变来源表位的研究发现,特异性识别该表位的TCR足以介导对表达该驱动突变抗原的肿瘤细胞的杀伤[38]。而在携带有KRASG12D突变的结直肠癌肺转移的患者中,通过从肺转移灶中分离出针对KRASG12D突变的肿瘤浸润T细胞,并在体外扩增回输入患者体内后,可引起10个肺转移灶中的9个病灶的消退[39]。这些都表明,P53和KRAS的驱动突变产生的肿瘤抗原可作为肿瘤治疗的有效作用靶点。
6 驱动突变抗原特异性T细胞受体工程化T细胞的肿瘤临床研究进展
目前,基于T细胞的免疫疗法已成功用于临床治疗人类实体瘤,TCR-T疗法正处于蓬勃发展中[40-45]。2006年,Science报道了一项通过逆转录病毒表达编码T细胞识别黑色素瘤抗原1(melanoma antigen recognized by T cells 1,MART-1)的特异性TCR,使来自外周血的自体T细胞获得了识别肿瘤的能力,进而让转移性黑色素瘤病变成功消退的临床研究[46]。然而,少数靶向肿瘤相关抗原的TCR-T治疗可引发严重甚至致死性的并发症,如结直肠癌TCR-T靶向癌胚抗原(carcinoma embryonic antigen,CEA)的临床治疗研究中并发了严重的炎性肠病而终止了研究[47]。靶向MART-1,糖蛋白100(glycoprotein 100,gp100)和黑色素瘤相关抗原A3(melanomaassociated antigen-A3,MAGE-A3)的黑色素瘤TCR-T治疗引起了正常组织的严重破坏[48]和严重的神经毒性反应致受试者死亡[49],以及心源性休克死亡的发生[50]。TCR-T疗法需要肿瘤特异性更强的抗原靶标,而针对驱动突变的TCR-T疗法由于驱动突变抗原的肿瘤专一表达,其可以有效降低肿瘤免疫治疗过程中的脱靶及细胞因子释放综合征和神经毒性的发生[51]。KRAS驱动突变是肿瘤的常见诱因,25%的非小细胞肺癌患者中发现存在KRAS第12和第13号密码子上的基因突变,而这些突变在结直肠癌患者中的发生率可达39%,在胰腺癌患者中的发生率更是高达95%[52]。Tran等[39]的发现表明KRAS驱动突变肿瘤新抗原可以在临床上有效诱导出T细胞免疫应答;而通过从肿瘤患者体内分离出的HLA亚型有效结合的肿瘤致癌突变抗原特异性T细胞,已筛选出多个识别KRAS驱动突变蛋白的特异性TCR序列。表1为截至2021年5月4日“https://clinicaltrials.gov”网站上已申报的关于驱动突变抗原及致癌病毒抗原肿瘤TCR-T疗法临床试验项目汇总,可见以特异性TCR为基础针对相应的肿瘤抗原开发TCR-T疗法在临床前和临床研究中显示出巨大的潜力。
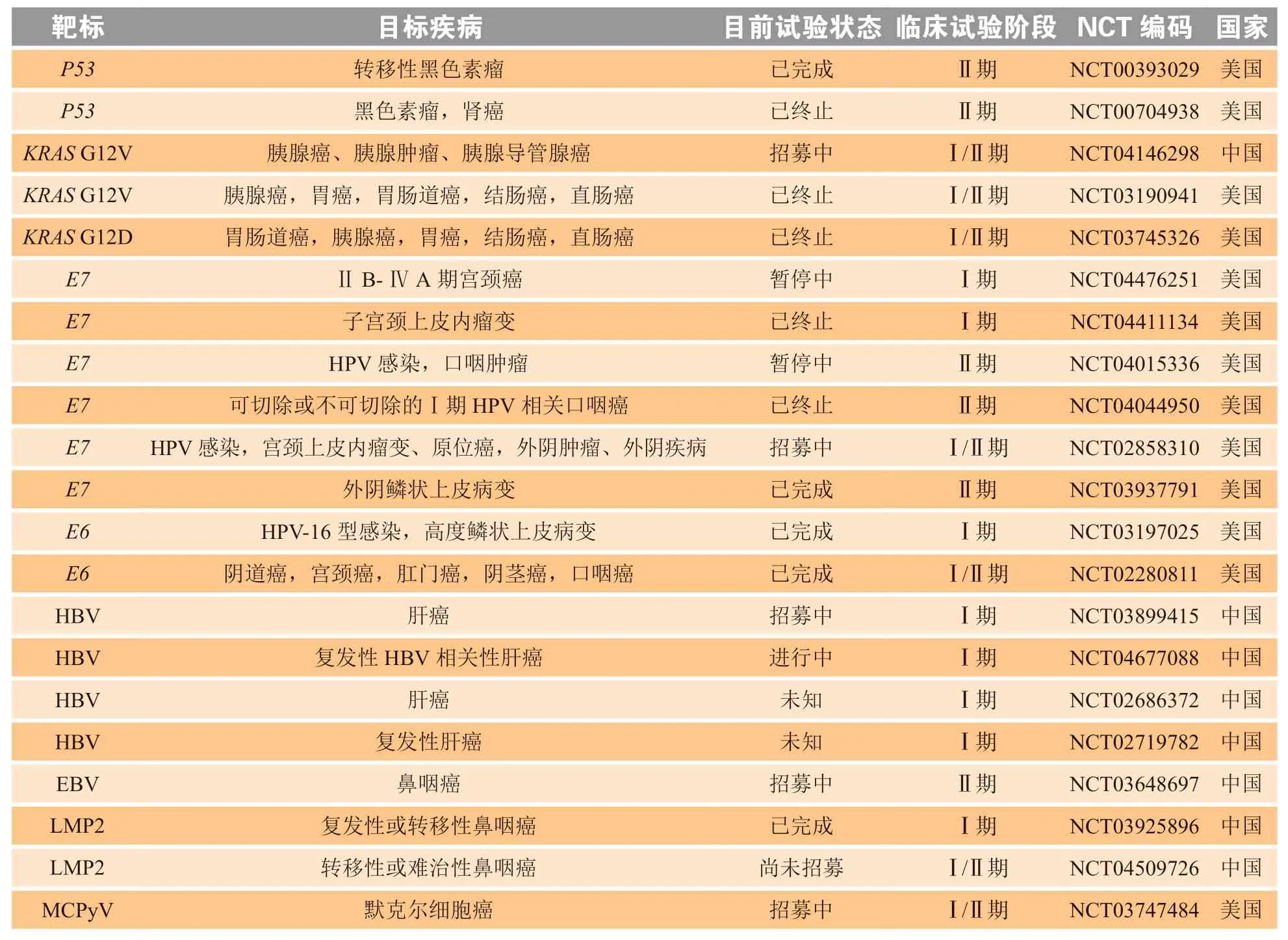
表1 驱动突变抗原及致癌病毒抗原肿瘤T细胞受体工程化T细胞疗法临床试验项目列表 Table 1 List of clinical trials of T cell receptor-engineered T cells therapy for driver mutation and oncogenic viral antigen related tumor
7 驱动突变抗原-T细胞受体筛选体系
7.1 基于活化T细胞核因子-绿色荧光蛋白报告基因的抗原-T细胞受体筛选系统
利用J76细胞系无内源TCR表达的特征,将候选功能性肿瘤抗原TCR重组并过表达于活化T细胞核因子-绿色荧光蛋白(nuclear factor of activated T cells-green fluorescent protein,NFATGFP)J76细胞可以构建抗原-TCR筛选系统(见图1A)。将上述报告细胞系与肿瘤抗原和DC共孵育,候选TCR若与对应抗原相互识别,则激活转录因子NFAT使其开始转录,其后的GFP表达并产生荧光信号,可通过流式细胞术定量检测[53-54]。由此,利用该系统,通过给予DC和不同肿瘤抗原孵育,可以从TCR组库中的候选TCR出发,在体外高效筛选可活化相应TCR克隆的肿瘤抗原。此方法相较于体内筛选更为简便,用时较少,结果更为显著。此功能报告基因系统也是其他高通量筛选方法的后续常用验证环节。
7.2 基于液滴微流控技术的功能性T细胞筛选系统
传统的单细胞分析方法依赖于与单细胞聚合酶链式反应(polymerase chain reaction,PCR)和高通量测序结合的流式细胞荧光分选(fluorescence activating cell sorter,FACS)技术,通常无法与实时的单细胞功能监测相提并论,并且受限于取样的细胞数而降低获得的T细胞的多样性。微流体领域的飞速发展为单细胞生物学的高通量研究提供了新的工具。其中,液滴微流控技术将单个细胞分离入极小体积的油滴,对单个细胞水平上细胞间的相互作用实时监测,并对其TCR序列进行下游识别,从而对T细胞进行功能筛选(见图1B)。这一液滴微流控平台包括针对每个单独克隆TCR的跟踪系统以及用于回收目标克隆以进行下游分析的分类程序。以识别NY-ESO-1抗原的T细胞为例,将T细胞与表达复杂HLA/NYESO-1抗原肽的靶细胞一起封装为单个液滴,非特异性T细胞(MART-1 T细胞)不识别表达NY-ESO-1抗原的靶细胞,而特异性T细胞(NY-ESO-1 T细胞)在识别其相关抗原后被激活,从而触发增强绿色荧光蛋白(enhanced green fluorescent protein,eGFP)的表达,经过液滴捕获、孵育和成像,观察eGFP的表达强弱,从而实现特异性T细胞的功能筛选[55]。该技术只需很少的细胞输入且整个样品被封装,从而大大减少了操作中的细胞损失,适用于珍贵且细胞数目有限的样品。此外,极小的液滴体积可有效提高灵敏度,从而减少分析时间;同时,其可对液滴中的单个细胞进行多参数检测和多功能筛选。
7.3 基于信号传导和抗原呈递双功能受体文库的抗原-T细胞受体筛选系统
T细胞识别靶抗原后被激活,进而诱导表达了可被检测到的基因,然而由于MHC分子缺乏信号结构域,对相应抗原提呈细胞(antigen presenting cell,APC)所提呈抗原的检测有一定挑战性。相关研究表明,采用信号传导和抗原呈递双功能受体(signaling and antigen-presenting bifunctional receptor,SABR)文库,可以高效识别与给定TCR相互作用的APC所呈递的抗原表位,对TCR靶向抗原的发现十分有效[56](见图1C)。SABR的细胞外结构域是共价连接的抗原肽-β2微球蛋白-MHC三聚体,参与提呈抗原,其与细胞内CD3ζ信号结构域和CD28共刺激结构域融合,可在递呈靶向抗原与TCR结合时向APC转导类似TCR的下游信号。在APC中导入NFAT-GFP报告基因即可获得具有抗原呈递报告功能的细胞系。基于SABR文库进行TCR-抗原筛选时,首先需生成由免疫表位数据库的所有已知HLA-A*0201限制性表位组成的目标表位列表,以编码抗原表位-SABR文库(A2-SABR文库),通过表位的蛋白质序列合成SABR文库的全部寡核苷酸;然后用免连接法将文库扩增并克隆至SABR载体质粒,通过慢病毒将后者转导至具有NFAT-GFP报告基因的JURKAT细胞的APC中,使其表达SABR。将此APC与具有可识别特定抗原TCR的JURKAT细胞共孵育,再利用流式细胞术分选GFP阳性的NFAT-GFP-JURKAT APC,并对所分选细胞的SABR抗原表位部分进行扩增和测序,与A2-SABR文库比对筛选获得确切抗原表位序列信息,由此成功进行特定TCR靶向抗原的高通量筛选。
7.4 基于T-Scan报告基因的高通量抗原-T细胞受体筛选系统
T-Scan采用基于细胞的联合筛选方法来高通量鉴定TCR有效识别的抗原(见图1D)。在T-Scan系统中,候选抗原文库通过慢病毒在靶细胞内表达,靶细胞加工后荷载到MHC分子上,在细胞表面通过MHC分子呈递出来。将这些带有抗原库的靶细胞与CD8+T细胞共同培养,TCR与抗原相互识别后,CD8+T细胞分泌细胞毒性颗粒至带有相应抗原的靶细胞。颗粒酶B(granzyme B,GzB)是一种存在于细胞毒性颗粒中的丝氨酸蛋白酶。靶细胞与CD8+T细胞成功识别后,分泌的GzB通过剪切连接红外荧光蛋白(infrared fluorescent protein,IFP)2个结构域间的GzB特异性剪切序列,激活靶细胞中的IFP,从而可以通过FACS分离出这些靶细胞。最后,使用PCR和二代测序(next generation sequencing,NGS)技术来鉴定这些靶细胞表达的抗原[57]。T-Scan不直接测量反应性T细胞,而是基于GzB检测CD8+T细胞对候选抗原库的聚焦作用,其允许从候选抗原库中富集具有高灵敏性和特异性的稀有同源靶细胞。T-Scan可以从蛋白质组中识别病毒和自身反应性TCR的已知肽表位,并继续利用该方法生成高分辨率的TCR特异性图谱,识别多个孤立TCR的对应表位。这种方法能够在高通量筛选中获得高度复杂的候选抗原组。
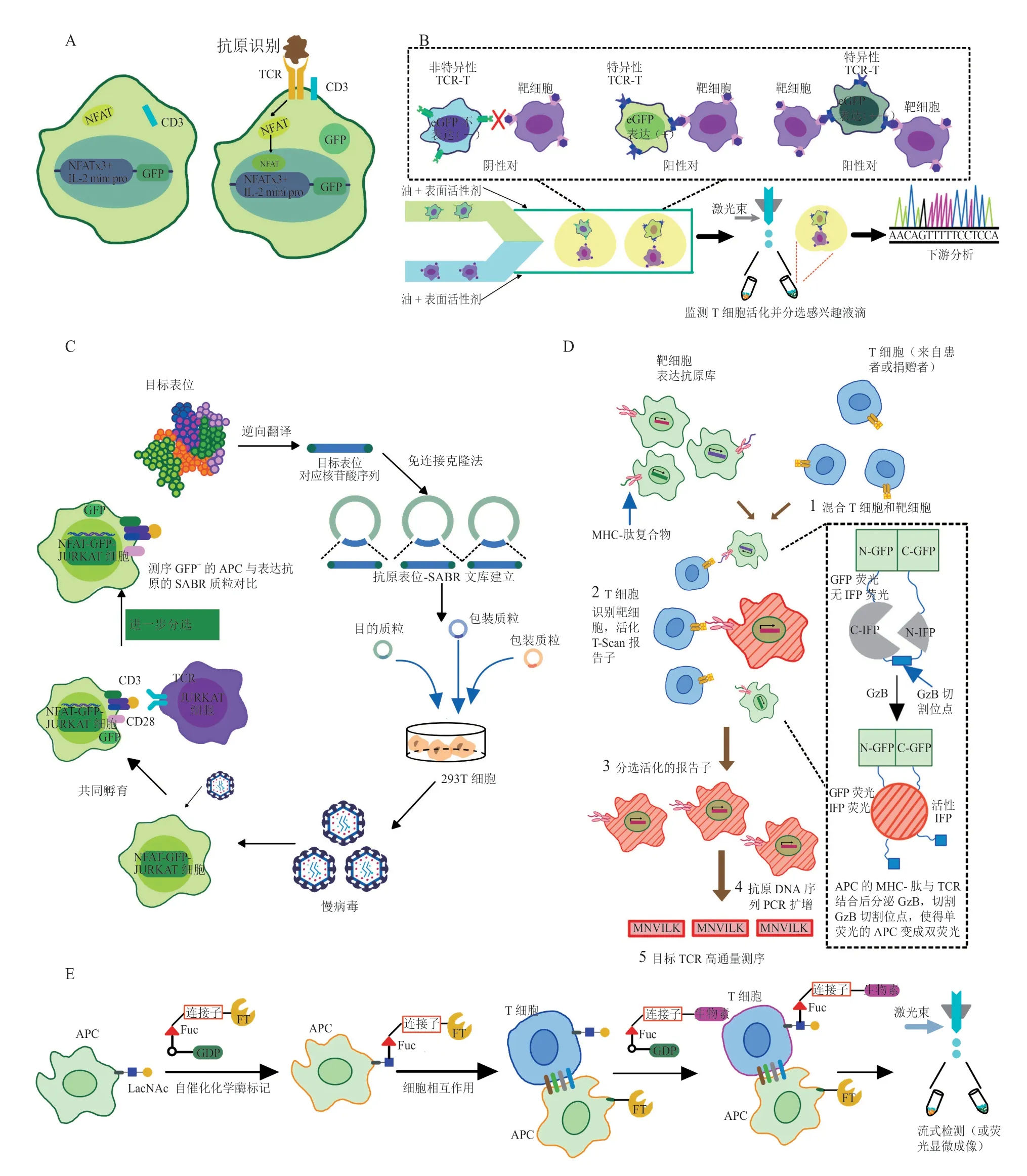
图1 抗原-T细胞受体筛选系统组图Figure 1 Charts of antigen-T cell receptor screening system
7.5 基于细胞接触和表面修饰的抗原-T细胞受体筛选系统
以细胞-细胞间相互作用为基础,对T细胞进行岩藻糖基-生物素化标记的抗原-TCR筛选系统可以检测并从旁观者T细胞中分离出针对肿瘤特异性抗原的反应性T细胞(见图1E)。岩藻糖基转移酶(fucosyltransferases,FT)是一种能快速定量地将与其天然供体底物鸟苷二磷酸岩藻糖(guanosine diphosphate-fucose,GDP-Fuc)结合的蛋白质转移到含有N-乙酰-D-乳糖胺(N-acetyl-D-lactosamine,LacNAc)的DC或T细胞表面的糖基转移酶。DC对肿瘤裂解物进行识别、加工和提呈表达肿瘤特异性抗原-MHC复合体;将人工合成的共轭化合物GDP-Fuc-FT与DC 共孵育,FT可作为自催化剂转移至DC表面;再将此DC与GDP-Fuc-生物素和T细胞共孵育,T细胞识别DC表面的肿瘤特异性抗原-MHC复合体,其特异性TCR与DC形成细胞-细胞间接触,DC表面的FT催化GDP-Fuc-生物素向T细胞表面转移并形成LacNAc-Fuc-生物素标记,通过亲和素荧光标记这一生物素化的T细胞后,可通过荧光显微成像或流式检测特异性识别肿瘤特异性抗原的T细胞[58]。此法不依赖遗传操作,较为简便,成本低,周期短,适用于原代细胞,通常适用于可检测到T细胞浸润的小鼠肿瘤模型。而在人的DC表面导入FT也容易完成,可用于检测和分离患者的肿瘤特异性抗原反应性T细胞,在临床中有很高的应用潜力,有望推动个性化癌症治疗的进一步发展。
8 展望及总结
在TCR-T治疗中,肿瘤特异性抗原及相对应的特异性TCR是肿瘤免疫应答的关键因素,更是免疫应答效应的分子基础。绝大多数的TCR-T疗法的靶标抗原特异性程度不足,使得治疗相关的副反应严重阻碍了这一疗法的发展。在早期T细胞靶向肿瘤突变抗原的研究中,所检测到的绝大部分肿瘤的突变负荷值较低,肿瘤细胞产生的能诱导免疫应答的肿瘤抗原极少;而致癌病毒抗原具有很强的免疫抗原性,是TCR-T治疗的理想靶点;驱动突变抗原是肿瘤细胞所特有的,并决定了肿瘤发生、发展的肿瘤特异性抗原,这一特性使得肿瘤驱动突变抗原自然而然成为TCR-T治疗的最优靶标,而这些驱动突变抗原及相应的TCR还有待进一步发掘,如通过对TCGA数据库的挖掘驱动突变基因,运用计算机预测肿瘤驱动突变抗原,通过肿瘤浸润细胞的单细胞RNA转录组和TCR联合测序获得潜在的功能性TCR信息,并通过TCR-pMHC晶体结构的解析等手段对驱动突变抗原和功能性TCR进行优化,还可以应用基于NFAT-GFP报告基因的抗原-TCR筛选系统、基于液滴微流控技术的功能性T细胞筛选系统等,这将极大推进实体瘤TCR-T治疗的发展。目前,TCR-T疗法正处于蓬勃发展之中,在临床前和临床研究中均展示出巨大的潜力。经过十几年的研究,其对于转移性黑色素瘤的治疗已被广泛采纳并进行了相关临床试验,而更多的针对实体瘤的TCR-T疗法也在积极的开展中。
