T细胞受体基因工程T细胞治疗的现状与未来
2021-11-13王丹阳熊昕昕刘海平周鹏辉
王丹阳,熊昕昕,刘海平,周鹏辉*
(1. 华南肿瘤学国家重点实验室 中山大学肿瘤防治中心,广东 广州 510060;2. 广东省人民医院 广东医学科学院,广东 广州 510055;3. 广州泛恩生物科技有限公司,广东 广州 510535)
根据GLOBOCAN 2020最新数据,2020年全球新增癌症病例达1 930万,并且有将近1 000万人死于癌症[1]。手术、传统化疗、放射治疗等传统治疗方法在癌症治疗中一直起着主导作用,然而这些方法的治疗效果较为局限,尤其是晚期患者很难从这些治疗方法中长期获益。近年来以基因修饰T细胞为中心的过继细胞治疗(adoptive cell therapy,ACT),包括嵌合抗原受体T细胞(chimeric antigen receptor T cell,CAR-T)和T细 胞 受 体 基因 工 程T细 胞(T cell receptor-engineered T cell,TCR-T)疗法[2],引起全世界广泛的关注,各种研究及临床试验数量呈指数级增长。其中,CAR-T疗法在血液肿瘤中取得了卓越的疗效[3-4],但是部分研究认为其在实体肿瘤上的治疗效果并不令人满意[5]。TCR-T治疗在实体肿瘤的治疗中具有非常大的发展潜力,本文阐述TCR-T治疗的优势、目前的研究现状以及现存的问题和挑战,并将讨论未来TCR-T治疗的发展方向,以期获得更好的治疗效果和更低的毒性或副作用,为实体肿瘤患者带来更高的获益。
1 嵌合抗原受体T细胞与T细胞受体基因工程T细胞
T细胞在癌症患者的适应性免疫应答中发挥着重要的作用,它通过T细胞受体(T cell receptor,TCR)识别特异性肽表位与主要组织相容性复合体(major histocompatibility complex,MHC)形成的复合物而被激活,这些肽表位主要来源于肿瘤细胞表达的内源性蛋白[6]。CAR-T是人工制造的可表达靶向肿瘤的嵌合抗原受体(chimeric antigen receptor,CAR)的T细胞,CAR是由单克隆抗体(monoclonal antibody,mAb)的单链可变片段(single chain variable fragment,scFv)与CD3 ζ链和1或2个共刺激信号基因融合而成的抗原受体[7]。TCR-T则是指将具有肿瘤抗原特异性的TCR通过基因工程技术导入到T细胞中,使T细胞获得识别和杀伤肿瘤细胞的功能[8]。CAR-T与TCR-T结构如图1所示。尽管CAR-T治疗对血液系统恶性肿瘤显示出良好的效果,但对实体肿瘤的效果并不令人满意[9]。
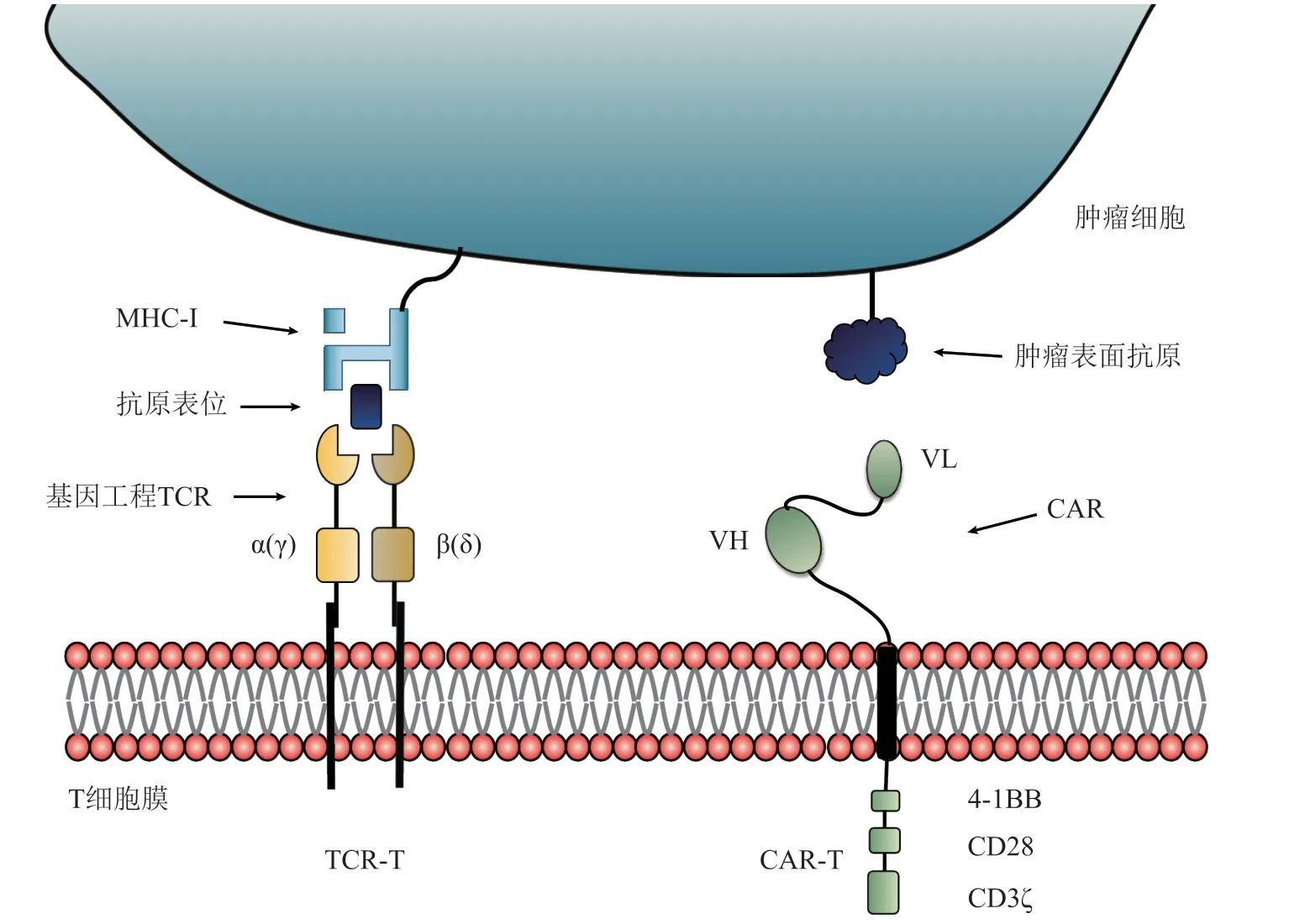
图1 T细胞受体基因工程T细胞与嵌合抗原受体T细胞Figure 1 T cell receptor-engineered T cell and chimeric antigen receptor T cell
与CAR-T相比,TCR-T具有多个优势,有望成为 ACT治疗实体瘤的新支柱。在CAR-T治疗中,CAR中的scFv识别的抗原通常为表达于细胞膜的蛋白。然而,膜蛋白的数量较为有限,在肿瘤细胞中特异表达但在正常组织中不表达或低表达者过少。因此,能够应用于实体瘤的CAR-T治疗靶点非常有限。而MHC呈递的抗原包含了细胞内表达的所有蛋白,无论胞内抗原、膜抗原还是分泌蛋白抗原,均有可能与MHC分子结合,从而被细胞的TCR识别,因此TCR-T治疗具有更广的靶标范围[10]。另外,部分肿瘤是由致癌病毒引起,靶向癌病毒抗原同样能够获得相应的治疗效果。而大部分病毒抗原属于胞内蛋白,因此TCR-T治疗也是病毒性肿瘤细胞治疗中的优选方案[11]。此外,每个CAR需要1 000余个抗原来激活,而每个TCR的激活仅需1 ~ 50个抗原,这使得TCR-T能够识别低丰度的肿瘤抗原[12]。另有研究表明,CAR-T主要分布在肿瘤周围以获取表面抗原,而TCR-T可以穿透肿瘤,这使得TCR-T在癌症治疗中更有效率[13]。
CAR-T治疗中出现的严重不良反应也限制了其发展,常见的不良反应有细胞因子释放综合征(cytokine release syndrome,CRS)、CAR-T相关脑病综合征(CAR-T related encephalopathy syndrome,CRES)等[14]。CRS主要由CAR-T与靶细胞结合后活化产生大量细胞因子和趋化因子,进而导致全身炎症反应所致,主要表现为高热,严重者可出现肺水肿、低血压、多器官衰竭和循环衰竭而死亡。尽管TCR比CAR的敏感性更高,但TCR-T治疗中的细胞因子释放量更少[10,15]。因此,与CAR-T治疗相比,TCR-T治疗发生CRS的风险可能更低。
2 肿瘤抗原
肿瘤免疫治疗的效果在很大程度上依赖于合适的肿瘤抗原和可特异性识别抗原的T细胞。从1991年首个人类肿瘤抗原——黑色素瘤抗原-A1(melanoma-associated antigen A1,MAGEA1)被鉴定以来[16],到目前已在各种肿瘤中发现了许多不同的肿瘤抗原,大致可分为肿瘤相关抗原(tumor associated antigens,TAAs)、肿瘤相关病毒抗原(tumor associated viral antigens)、肿瘤特异性抗原(tumor-specific antigens,TSAs)、非传统的肿瘤抗原(unconventional tumor antigens,UCAs)4类[17]。
2.1 肿瘤相关抗原
TAAs是指在某些正常组织中低表达或在免疫豁免器官中表达,但在肿瘤中高表达的抗原,是正常基因的产物。如癌睾丸抗原(cancer-testis antigen,CTA)、癌胚抗原(carcinoembryonic antigen,CEA)和组织分化抗原(tissue differentiation antigens,TDAs)等。纽约-食管癌抗原-1(New York esophageal squamous cell carcinoma 1,NY-ESO-1)和MAGE-A属于在睾丸中特异表达,而在某些肿瘤组织中异常表达的CTA;CEA在胚胎发育过程中表达,胚胎发育完成后停止表达,但在一些肿瘤组织中出现异常高表达,目前被认为是结直肠癌和其他一些肿瘤的重要标志物;TDAs则通常局限于肿瘤和肿瘤来源的组织,例如糖蛋白100(glycoprotein 100,gp100)局限于皮肤、前列腺特异性抗原(prostate specific antigen,PSA)局限于前列腺等;另外还有一部分在癌症中过表达的肿瘤抗原,例如人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)和 人 内源性逆转录病毒(human endogenous retroviruses,HERVs)抗原。HERVs是几百万年前的逆转录病毒整合到人类基因组中的残余物,在正常组织中是表观遗传沉默的,但可以通过DNA去甲基化在肿瘤中表达,成为抗原。目前针对TAAs的TCR-T治疗已有多个临床试验在开展。
2.2 肿瘤相关病毒抗原
一部分实体瘤因病毒感染而产生,其中病毒基因整合到细胞基因组导致具有致癌特性的病毒蛋白的表达。例如,E6和E7癌蛋白的表达是由感染人类乳头状瘤病毒(human papillomavirus,HPV)的高危毒株产生,它促进了一些头颈部癌、宫颈癌和肛门癌亚群的发生和发展[18]。此外肝癌的发生与乙型肝炎病毒和丙型肝炎病毒有关,而鼻咽癌与EB病毒(Epstein-Bar virus,EBV)感染有关。病毒抗原与正常细胞蛋白显著不同,能够引发高亲和力的TCR反应,因此病毒抗原具有较好的特异性和免疫原性,也是TCR-T和CAR-T治疗的靶标抗原。
2.3 肿瘤特异性抗原
TSAs是由肿瘤细胞中的基因突变产生的新抗原,也被称为新生抗原(neoantigens)。这类抗原在正常组织中不存在,因此针对这类抗原的免疫治疗对正常组织的毒性很小。另外,这些抗原也没有参与胸腺中的T细胞阴性选择,因此与TAAs相比,体内存在识别TSAs的高亲和力TCR,更适合进行免疫治疗。然而,TSAs来源于基因组中的随机突变,大部分驱动基因突变的免疫原性低,因此针对这类抗原的TCR-T只适合个体化治疗。另外,TSAs既要有一定的表达量,还必须与MHC分子有效结合才能够被呈递到细胞表面成为抗原,因此只有一部分突变的蛋白能够成为肿瘤抗原。目前,在新生抗原的预测和鉴定上仍存在较大的问题,预测的准确率较低。同时,筛选和鉴定识别某个抗原的特异TCR也需要较长的时间。以上原因导致靶向特定新抗原的TCR-T治疗难以实现,以TSAs为基础的肿瘤疫苗则有多个临床试验在开展。鉴于患者的免疫系统在抗肿瘤免疫反应中会激活识别TSAs的T细胞,而这些肿瘤抗原特异性T细胞(tumor antigen-specific T cells,Tas)所携带的TCR可特异识别TSAs,因此如果能够特异分离Tas并克隆这些细胞的TCRs,则能够快速制备出TSAs特异的TCR-T,进行个体化治疗,并且无需鉴定TSAs的抗原序列。
2.4 非传统的肿瘤抗原
UCAs是指通过异常转录、翻译或翻译后修饰,从基因组的非编码区域或编码区域产生的抗原。这些过程可能不完全是肿瘤特异性的,也可以发生在正常组织中。因此,非传统的抗原可能具有TAAs的性质,也可能具有TSAs的性质。
3 T细胞受体基因工程T细胞治疗的临床研究进展
目前,有大量针对不同抗原的TCR-T治疗进入临床试验(见表1)。已有的临床试验结果表明,TCR-T治疗是一种有效的肿瘤治疗方法。Johnson等[19]的研究显示,接受T细胞识别黑色素瘤抗原1(melanoma antigen recognized by T cells 1,MART-1)和gp100 TCR-T治疗的黑色素瘤患者的客观有效率分别为30%和19%,并且在细胞回输1个月后,所有患者的血液中仍保持了高水平的TCR-T。Rapoport等[20]的研究显示,20例晚期多发性骨髓瘤患者接受NY-ESO-1 TCR-T治疗,其中有16例(80%)观察到了治疗效果。在另外一项NY-ESO-1 TCR-T治疗转移性黑色素瘤和滑膜细胞肉瘤患者的临床研究中,患者出现了持久的反应[21-22]。近期的一项NY-ESO-1 TCR-T治疗复发、难治性或高危多发性骨髓瘤的研究显示,细胞治疗的中位总生存期为35.1个月,其中2例患者在近5年后病情无进展[23]。Tawara等[24]靶向Wilms瘤基因1(Wilms tumor gene 1,WT1)的TCR-T治疗研究中,8例患者被分为2个剂量组接受TCR-T治疗,结果均未观察到靶向正常组织的不良事件;在研究结束时,5例患者可持续检测到TCR-T,并且这5例患者中有4例存活超过12个月。这些结果均反映了TCR-T治疗的有效性。
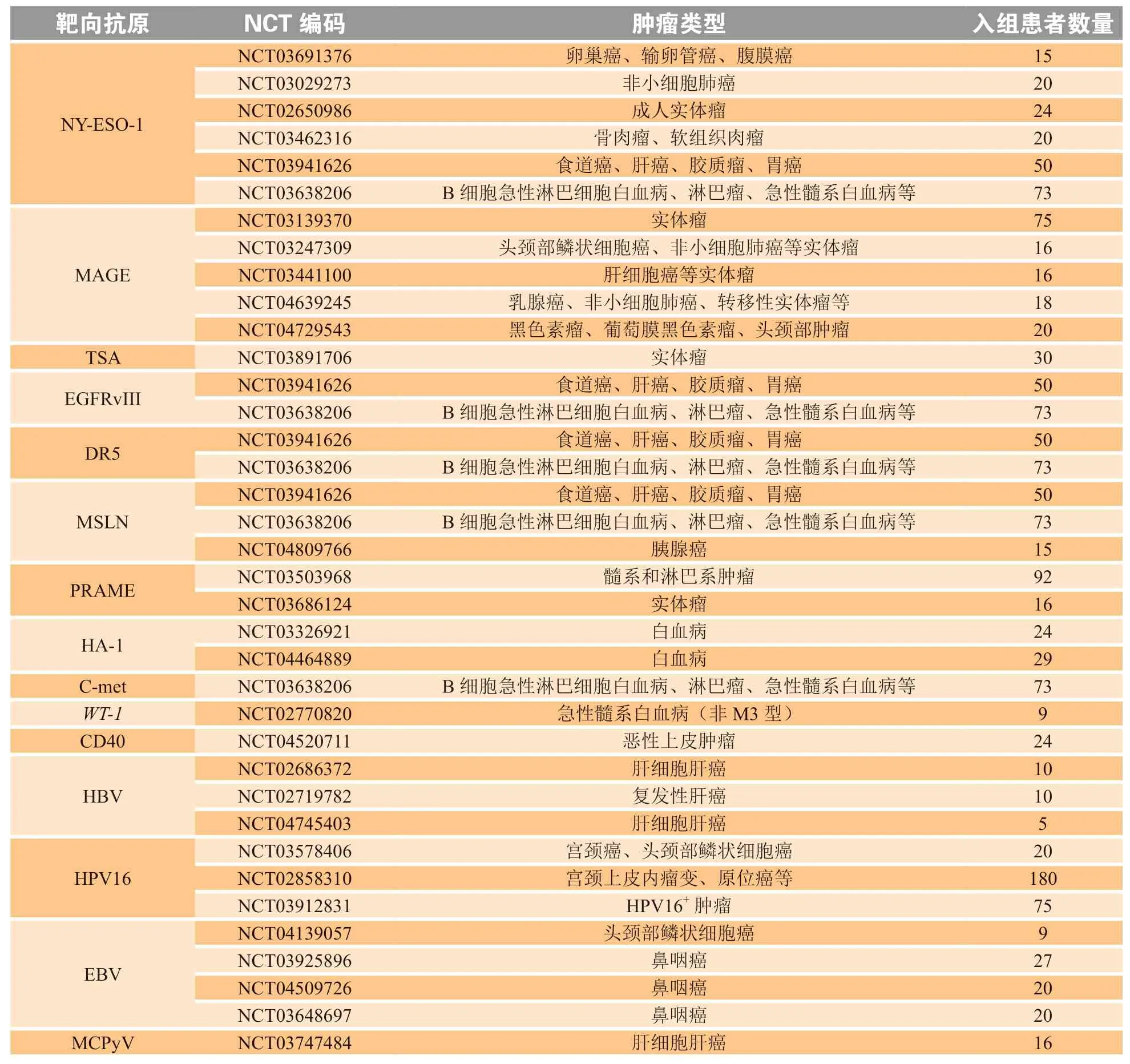
表1 T细胞受体基因工程T细胞治疗临床试验Table 1 Clinical trials of T cell receptor-engineered T cell therapy
4 T细胞受体基因工程T细胞治疗面临的挑战
4.1 肿瘤靶向的瘤外毒性
当TCR-T靶向的抗原不只在肿瘤上表达时,就会产生肿瘤靶向的瘤外毒性(on target off tumor toxicity)。Parkhust等[25]在转移性结直肠癌中使用了以CEA作为靶点的高亲和力TCR-T进行治疗,尽管所有接受治疗的患者血清中的CEA水平均下降,但有3例患者出现了危及生命的结肠炎和结肠出血,试验被迫终止。一项以MAGE-A3为靶点的TCR-T治疗临床试验中,9例MAGE-A3高表达的患者(包括转移性黑色素瘤、滑膜肉瘤和食管癌)中5例患者在治疗后获得了局部反应;2例患者由于脑组织中表达的MAGE-A12含有与MAGE-A3类似的肽序列,导致TCR-T在大脑中产生交叉反应,出现严重的脑灰质损伤,最后死亡[26]。
4.2 T细胞抗原受体α和β链的错配及外源T细胞抗原受体的表达降低
大多数T细胞表达αβTCR,该TCR由肽链α和β组成,少数T细胞表达γδTCR,该TCR由肽链γ和δ组成[27]。目前的TCR-T多为αβTCR。将外源性TCR引入T细胞后,外源性TCR的α和β链与内源性TCR的α和β链同时存在于同一T细胞中,会产生外源与内源α和β链之间的错配[28]。对接受TCR-T输注小鼠的研究表明,这种错配出现的新型TCR可能会导致严重的移植物抗宿主病(graft versus host disease,GVHD)[29]。此外,外源TCR必须与内源TCR以及错配的TCR竞争在细胞表面的表达,包括与CD3分子的竞争结合,这可能导致外源TCR在膜表面的表达水平降低,无法形成有效的T细胞激活,降低肿瘤杀伤功能[30]。因此,必须采取措施防止TCR的错配。
4.3 主要组织相容性抗原复合体限制性
由于每个TCR均有其对应的MHC类型,导致患者既要表达靶向抗原,又要表达相应的MHC分子,才能进行治疗。这使得适合TCR-T治疗的患者数量大大减少。人类白细胞抗原(human leukocyte antigen,HLA)是人类的MHC表达产物,其中HLA-A*0201亚型是白种人中的高频HLA分子[31],现有的TCR-T临床试验大多数与此亚型相关,而针对其他HLA亚型的TCR-T治疗很少。
4.4 肿瘤微环境对T细胞受体基因工程T细胞的抑制
实体瘤微环境通过多种复杂的机制抑制T细胞介导的抗肿瘤免疫反应,包括减少T细胞的浸润、诱导T细胞耗竭等。TCR-T同样面临相同的问题。通过静脉回输的TCR-T需要高效进入肿瘤组织才能够产生较强的抗肿瘤免疫反应,从而获得治疗效果。研究显示,进入肿瘤的TCR-T长期暴露于肿瘤抗原刺激,同样会发生T细胞耗竭,导致TCR-T功能失调,降低治疗效果[32]。
4.5 T细胞受体基因工程T细胞的制备
晚期肿瘤患者的疾病进展迅速,过长的TCR-T制备流程会导致患者无法及时获得治疗,降低治疗效果。另外,晚期肿瘤患者的外周免疫细胞功能状态较差,影响TCR-T制备的成功率。目前,在较短时间内制备出高质量的TCR-T是一个挑战[33]。
5 T细胞受体基因工程T细胞治疗的未来
5.1 靶向肿瘤特异性抗原的T细胞受体基因工程T细胞治疗
由于TSAs来自肿瘤细胞中的随机突变,无法提前制备好相应的TCR-T,因此目前TCR-T治疗均是靶向TAAs[34]。但是TAAs的数量非常有限,并且无法克服TAAs在正常组织中表达所引起的毒性和副作用。由于TSAs具有较高的免疫原性,且在正常组织中表达缺乏,因此靶向TSAs在疗效和安全性上均具有显著的优势。传统的TCR-T的研制流程通常是在明确抗原序列后,通过筛选获得特异识别抗原的TCR,并明确对应的MHC亚型再应用于治疗。整个过程耗时长,效率低,不适合TSAs这类个体化的抗原。然而,患者的抗肿瘤免疫反应会激活自身体内识别TSAs的T细胞,例如既往已经从肿瘤患者中鉴定了识别TP53突变和KRASG12V突变的T细胞和TCR[35-36]。因此,可以从患者自身的肿瘤抗原特异T细胞中快速获得特异识别TSAs的TCR,用于制备个体化的TCR-T进行治疗。笔者团队通过鉴定肿瘤抗原特异T细胞的分子标志物,实现了从患者的肿瘤组织中准确分离肿瘤抗原特异T细胞,并快速克隆其TCR,建立了个体化TCR-T治疗的全套技术平台,并开始了Ⅰ期临床研究(NCT03891706)。
5.2 避免T细胞抗原受体的错配
Morton等[37]通过规律成簇的间隔短回文重复(clustered regularly interspaced short palindromic repeat,CRISPR)/CRISPR相关核酸酶9(CRISPRassociated nuclease 9,Cas9)编辑TRAC和TRBC基因位点,实现了内源性TCR的α和β链的基因敲除,从而增加了外源TCR的表达和功能,并在多发性骨髓瘤的临床前模型中证实,增加外源TCR的表达可以增强TCR-T对目标抗原的识别,延长对肿瘤生长的抑制。另有研究显示,将αβTCR导入到γδT细胞中能够显著减少TCR的配对错误[38]。而将γδTCR转导到αβT细胞同样不会出现2种TCR之间的错配。He等[39]制作了表达TCRγ4δ1的αβT细胞,发现这类T细胞不表达混合的TCR二聚体,也不结合或杀伤正常细胞,并在人源肝癌肿瘤细胞HepG2裸鼠模型中显著抑制了肿瘤生长。
5.3 通用型T细胞受体基因工程T细胞
通过敲除HLA分子,降低宿主免疫系统的排斥,敲除异体T细胞的内源TCR避免对宿主组织的免疫攻击,可以制备出通用型的T细胞用于TCR-T治疗,这不仅可以提高产品的一致性,缩短制备时间,还能够解决晚期患者自体T细胞质量不足的缺点,是未来T细胞治疗的重要发展方向[40]。
5.4 提高T细胞受体基因工程T细胞的肿瘤浸润效率
在实体瘤中产生疗效的前提是有足够的TCR-T进入到肿瘤组织中发挥作用。而肿瘤为了逃逸免疫攻击,其内部环境包括血管结构与状态并不适合TCR-T的浸润,如帮助效应T细胞迁移的趋化因子和黏附分子等的表达水平低,从而降低了TCR-T进入肿瘤组织的效率[41]。在CAR-T中,表达CXCR2等趋化因子受体可以改善T细胞向肿瘤组织的转运迁移[42-43],类似的方法也可以应用到TCR-T中。
5.5 抵抗肿瘤微环境抑制
肿瘤微环境通过多种复杂机制抑制T细胞功能。通过干预T细胞内部的抑制信号通路,分泌因子改善肿瘤微环境等方式,能够有效提高TCR-T的治疗效果。在T细胞中直接敲除抑制性受体如程序性死亡受体-1(programmed cell death protein 1,PD-1)的基因等,可降低T细胞在肿瘤微环境中的耗竭[44]。另外,临床前研究结果显示,在抗PD-1抗体存在的情况下,HER2 CAR-T对肿瘤的生长抑制有显著改善,并且该治疗效果与PD-1阻断后HER2 CAR-T功能增加相关[45]。这或可为TCR-T与免疫检查点抑制剂的联用提供思路和佐证,当然,疗效和副作用也需要有更多的临床前研究和临床试验进一步探讨。
综上所述,TCR-T治疗目前处于一个早期并快速发展的阶段,其已在临床中表现出较好的治疗效果,是肿瘤免疫治疗领域的重要发展方向之一。未来随着相关技术的发展和瓶颈的突破,以及个体化TCR-T治疗技术的建立,TCR-T治疗将成为肿瘤治疗的重要手段。
