T细胞受体工程化T细胞疗法的临床研究及动物肿瘤模型的应用
2021-11-13王琨汪金姣王皞鹏
王琨,汪金姣,王皞鹏
(上海科技大学生命科学与技术学院,上海201210)
过继性细胞疗法(adoptive cell therapy,ACT)在癌症治疗方面具有巨大的应用价值,它通过基因修饰技术对天然T细胞进行改造,使它们能够特异性识别肿瘤以获得肿瘤杀伤能力[1]。ACT主要包括肿瘤浸润淋巴细胞(tumor infiltrating lymphocytes,TILs)疗法、自然杀伤细胞(natural killer cell,NK)疗法、T细胞受体工程化T细胞(T cell receptor-engineered T cells,TCR-T)疗法和嵌合抗原 受 体T细 胞(chimeric antigen receptor T cells,CAR-T)疗法等。目前,CAR-T和TCR-T疗法应用最广。
CAR-T疗法已被证实可用于复发或难治性B细胞淋巴瘤如急性淋巴细胞白血病、弥漫性大B细胞淋巴瘤和多发性骨髓瘤的治疗[2-4]。目前,美国FDA总共批准了5款CAR-T治疗产品,为肿瘤患者带来了新的希望。尽管CAR-T疗法在血液瘤的治疗中显示出卓越的疗效,但对于实体瘤的治疗效果却不尽如人意。CAR-T疗法通过识别膜表面抗原而发挥作用,而近90%的恶性实体肿瘤缺乏膜表面特异性抗原,TCR-T疗法则能够识别细胞内来源的肿瘤特异性抗原。这也意味着,TCR-T疗法拥有更多的肿瘤潜在靶点选择,能更好地减少免疫逃逸,最终更有效地实现肿瘤细胞的特异性免疫应答,达到精准治疗的目的。因此,目前TCR-T疗法是最有可能在实体瘤治疗中取得突破的T细胞免疫疗法。本文将聚焦于现有TCR-T疗法相关的非临床研究动物模型以及TCR-T疗法临床发展现状,并进一步探讨了动物模型在克服TCR-T疗法当前限制性因素中的相关应用,为提高其临床治疗效果提供高效且广谱的解决方案。
1 T细胞受体工程化T细胞疗法简述
TCR-T疗法是一种通过工程化改造T细胞,从而实现对肿瘤患者进行治疗的过继性免疫疗法。TCR-T疗法通过直接赋予T细胞特异性识别结合肿瘤抗原的TCR,使得原本无肿瘤识别能力的T细胞能够有效地识别并杀伤肿瘤细胞,治疗的基本步骤为:首先,从患者的外周血中分离纯化出T细胞;继而利用基因工程技术,将识别肿瘤特异抗原的TCR基因序列导入患者自身T细胞中,获得特异识别肿瘤抗原的TCR-T;之后,将TCR-T通过体外培养进行大量扩增;最后,扩增得到的TCR-T被回输到患者体内对肿瘤细胞进行杀伤,从而达到治疗肿瘤的目的。细胞内在的抗原递呈机制使TCR-T可针对细胞胞内抗原(占全部抗原的90%)而不局限在细胞表面抗原(仅占10%)。因此,TCR-T可以靶向大部分的肿瘤特异性或相关性抗原,这使其对肿瘤的识别范围比抗体类药物以及依靠抗体识别肿瘤的CAR-T的应用范围更广。TCR-T依赖天然的TCR发挥作用,意味着其抗原亲和力以及信号强度都要弱于CAR-T,这使得TCR-T的持久性和浸润性要优于CAR-T,从而在实体瘤的治疗中优于CAR-T疗法。
TCR-T疗法的出现要早于CAR-T疗法。20世纪90年代,来自美国国家癌症研究所(NCI)和华盛顿大学的研究人员最早提出了T细胞的治疗用途,首次从人外周血中分离出抗原特异性T细胞,并过继性回输给肿瘤或病毒感染的患者体内,取得了一定的临床治疗效果[5]。多项临床试验表明,体外扩增的TILs可用于治疗人类癌症,如恶性黑色素瘤[6-7]或卵巢癌[8]。NCI研究人员对TILs靶向肿瘤的治疗潜力进行了验证,结果显示该方法在72%的转移性黑色素瘤患者中产生了客观反应[9]。然而,TILs对其他恶性肿瘤的治疗成功率非常有限,主要原因是大多数癌症患者体内很难被分离出肿瘤特异性T细胞,另外获得足够数量T细胞需要相当长的时间。正是这些原因推动了TCR-T技术的发展,使得产生抗原特异性T细胞成为可能。此后,TCR-T疗法在转移性黑色素瘤、结直肠癌、滑膜肉瘤和多发性骨髓瘤患者中均显示出良好的临床效果,表明TCR-T疗法在实体瘤的治疗中具有很好的应用前景[10-14]。
2 T细胞受体工程化T细胞疗法非临床研究 动物模型
为了验证和优化TCR-T疗法在临床应用中的安全性和有效性,研究人员需要建立非临床研究的动物模型,并对治疗效果进行验证和评估。在利用TCR-T疗法进行治疗的过程中,存在的安全性问题或不良反应主要包括:靶标毒性(on-target toxicity),指TCR-T攻击肿瘤细胞的同时也攻击表达TCR-T靶向抗原的正常细胞,使得健康组织受损;脱靶毒性(off-target toxicity),指TCR-T无法区分肿瘤细胞表面特异性抗原和正常细胞抗原,损伤了表达与靶点相似抗原表位的健康组织;细胞因子释放综合征(cytokine release syndrome,CRS),指工程化的T细胞完成输注后,使得T细胞被激活并快速增殖,引起细胞因子的过度级联释放,是一种严重的过度免疫应答。建立合适的临床前治疗模型,可以加快TCR-T疗法的开发和应用,最大程度避免临床试验中可能出现的安全问题。目前,在TCR-T疗法的研究中主要使用2种啮齿类动物模型:一是同基因型小鼠模型,使用小鼠T细胞和小鼠抗原,具有保留着完整免疫系统的优点;二是异种移植瘤小鼠模型,使用免疫缺陷型小鼠和人源T细胞及人源肿瘤细胞,具有能够研究人源细胞的优点[15-16]。此外,在CAR-T疗法的研究中,灵长类动物模型也被用于评估该疗法的有效性和安全性。例如,在分别以ROR1,L1CAM以及CD20等肿瘤抗原为靶点的CAR-T临床前研究中,均使用了灵长类动物作为模型,它们为这些CAR-T产品的安全性提供了有价值的信息[17-19]。相比小鼠模型,灵长类动物的免疫系统与人类更相似,其模型更接近患者体内的情况。灵长类动物模型在CAR-T疗法中的应用对于TCR-T临床前治疗模型的构建具有很大的参考价值,相信未来会有更多种类的动物模型在TCR-T疗法临床前研究中得到应用。
随着TCR-T疗法的不断发展,已有多种用于研究的小鼠肿瘤模型被成功构建,包括表达T细胞识别的黑色素瘤抗原1(melanoma-associated antigen recognized by T cells 1,MART-1)的 黑 色素瘤模型[20-21]、表达纽约食管鳞状细胞癌1(New York esophageal squamous cell carcinoma 1,NYESO-1)抗原的小鼠肿瘤模型[22]、表达糖蛋白100(glycoprotein 100,gp100)抗原的小鼠肿瘤模型[23]、表达癌胚抗原(carcinoembryonic antigen,CEA)的结肠癌小鼠模型[24]、表达p53抗原的骨肉瘤小鼠模型[25]、爱泼斯坦-巴尔二氏病毒(Epstein-Barr virus,EBV)相关的小鼠肿瘤模型[26]、人乳头瘤病毒(human papilloma virus,HPV)相关的头颈部鳞状细胞癌小鼠模型[27]、乙型肝炎病毒(hepatitis B virus,HBV)相关的肝癌小鼠模型[28]等。研究人员在构建MART-1,NY-ESO-1,p53等肿瘤模型时,一般先将3×106~ 5×106个肿瘤细胞通过皮下注射的方式种植在免疫缺陷型小鼠皮下,而gp100,CEA,EBV,HPV,HBV等模型的肿瘤细胞种植数目相对较少,一般在5×105~ 10×105个细胞左右。模型构建的下一步需要根据肿瘤生长状况选择合适的时间点,将工程化改造后的肿瘤特异性TCR-T通过尾静脉注射的方式转移至小鼠模型体内。MART-1,NY-ESO-1,EBV等模型一般会在移植肿瘤细胞3 ~ 5周后输入TCR-T,而gp100,CEA,p53,HPV,HBV等模型会在移植肿瘤细胞1 ~ 2周后静脉注入TCR-T。此外,不同的肿瘤模型需要输入的TCR-T数目也会存在差异。MART-1,NY-ESO-1,gp100,HBV等肿瘤模型一般在输入1×106~ 3×106个TCR-T后,便会达到良好的肿瘤杀伤效果,而CEA,p53,EBV,HPV等模型一般需要静脉注入5×106~ 10×106个TCR-T才能达到理想的体内抗肿瘤功效。在构建肿瘤模型以及体内试验的整个过程中,需要对肿瘤的生长状况进行定期监测,最常用的2种监测方式是活体成像和游标卡尺测量肿瘤。目前,业内尚未建立起动物模型和临床治疗之间的TCR-T回输剂量关系。临床上在对患者进行TCR-T治疗时,回输细胞数目主要依据患者的体质量来确定,相较于CAR-T,TCR-T回输的数量要高100 ~ 1 000倍,大多集中在109~ 1010个细胞之间。此外,TCR-T疗法的回输剂量标准尚未建立,并且因细胞回输剂量过大而引起细胞毒性现象的研究少见。与CAR-T疗法相比,TCR-T疗法引起CRS的程度更低,但若是过量的T细胞回输后导致T细胞激活并快速增殖必然会引起细胞因子过度级联释放。因此,未来可以参考CAR-T疗法的剂量爬坡试验评估TCR-T疗法的临床安全性和有效性。
构建小鼠模型开展临床前相关研究是实现基础研究转化至临床应用的重要桥梁。临床数据显示,在接受CEA特异性TCR-T治疗的患者中观察到的细胞毒性与在CEA小鼠模型中观察到的毒性高度相似,TCR-T除了发挥抗肿瘤功能外,还破坏了正常的结肠组织,类似于自身免疫性结肠炎[29]。在Pmel-1TCR转基因小鼠模型中,使用gp100特异性TCR-T治疗后,引起了小鼠的眼部损伤,这与接受gp100特异性TCR-T治疗的黑色素瘤患者中出现的状况基本相似[30]。以上结果表明,在进行TCR-T疗法临床试验前,小鼠肿瘤模型构建的必要性。动物模型的构建能够评估TCR-T疗法的治疗潜力,预测TCR-T疗法的细胞毒性,为临床试验的安全有效进行提供保障。
3 T细胞受体工程化T细胞疗法临床研究现状
2006年,Morgan等[10]首次报道了针对黑色素瘤患者的TCR-T疗法。研究人员使用RNA电穿孔的方法给患者的外周血单核细胞(peripheral blood mononuclear cell,PBMC)转 导 了4种编 码TCR的RNA,它们分别能够识别MART-1:27-35,gp100:209-217,NY-ESO-1:157-165和p53:264-272肽/人类白细胞抗原A2(human leukocyte antigen A2,HLA-A2)复合物;依据实体瘤反应评估标准(RECIST)评估后,发现经过治疗的17例黑色素瘤患者对TCR-T疗法产生了不同程度响应,其中2例表现出转移性黑色素瘤的持续客观消退,1例在接受治疗后出现了腋下肿块的完全消退,肝脏肿块也减少了89%,治疗后第21个月,该患者仍未表现出疾病进展;另1例患者的肺门肿块出现消退,治疗后第20个月仍无疾病进展。Johnson等[11]使用高亲和力的MART-1(AAGIGILTV)特异性TCR治疗20例转移性黑色素瘤患者,其中6例(30%)出现了客观的癌症消退,肺部、脑部、肝部和皮肤的肿瘤明显缩小。
Robbins等[13]报道的靶向滑膜细胞肉瘤和黑色素瘤抗原NY-ESO-1的TCR-T疗法的临床试验结果显示,4例(67%)滑膜细胞肉瘤患者和11例(45%)黑色素瘤患者均出现了客观临床反应,其中有2例黑色素瘤患者表现出完全消退,1例滑膜细胞肉瘤患者表现出持续18个月的部分反应。2015年,Robbins等[14]报道了一项使用亲和力增强的TCR识别NY-ESO-1(SLLMWITQC)的临床试验结果:将TCR通过逆转录病毒转导入了18例滑膜细胞肉瘤患者和20例黑色素瘤患者的PBMC中,18例NY-ESO-1阳性滑膜细胞肉瘤患者中的11例(61%)和20例NY-ESO-1阳性黑色素瘤的患者中的11例(55%)表现出了客观的临床反应。
Parkhurst等[31]报道了针对结肠癌患者的TCR-T疗法临床试验结果,该项试验使用了靶向结肠癌高表达抗原CEA(一种糖基化蛋白,在多种胃肠道癌细胞中高表达)的TCR(IMIGVLVGV),临床结果显示,对其他疗法无效的3例转移性结直肠癌患者接受了TCR-T疗法后,均检测到CEA水平的显著降低,且1例患者肺和肝转移性肿瘤客观消退。Kageyama等[32]报道了靶向黑色素瘤抗原家族A4(melanoma antigen family A4,MAGE-A4)(NYKRCFPVI)的TCR-T疗法在10例对于常规治疗手段如放疗和化疗等响应较差或不响应的食管癌复发患者中的临床试验结果,这些患者在接受治疗后短期内肿瘤缩小,3例病情最轻微的患者在未接受其他治疗的情况下,1年后疾病不再进展。Nagarsheth等[33]报道了靶向HPV的TCR-T疗法临床试验结果:与HPV相关的恶性肿瘤是典型的上皮癌,包括宫颈癌、口咽癌、肛门癌、外阴癌、阴道癌和阴茎癌均表达HPV E7抗原,有助于恶性转化和癌细胞存活,HPV E7抗原可作为工程T细胞的治疗靶点;12例转移性HPV-16阳性癌症患者中8例曾接受程序化细胞死亡蛋白1(PD-1)免疫治疗、1例曾接受LN-145细胞治疗,效果均不理想,接受靶向TCR的HPV-16 E7工程改造的T细胞(E7 TCR-T)治疗后,6例出现了明显的肿瘤消退。
综上所述,在这些TCR-T疗法的临床试验中,针对同种肿瘤的不同试验的临床客观性也会出现差异,这与针对同一抗原的不同TCR的选择和TCR-T制备工艺之间的差异有关。表1显示了目前已完成的关于TCR-T疗法的临床试验结果。
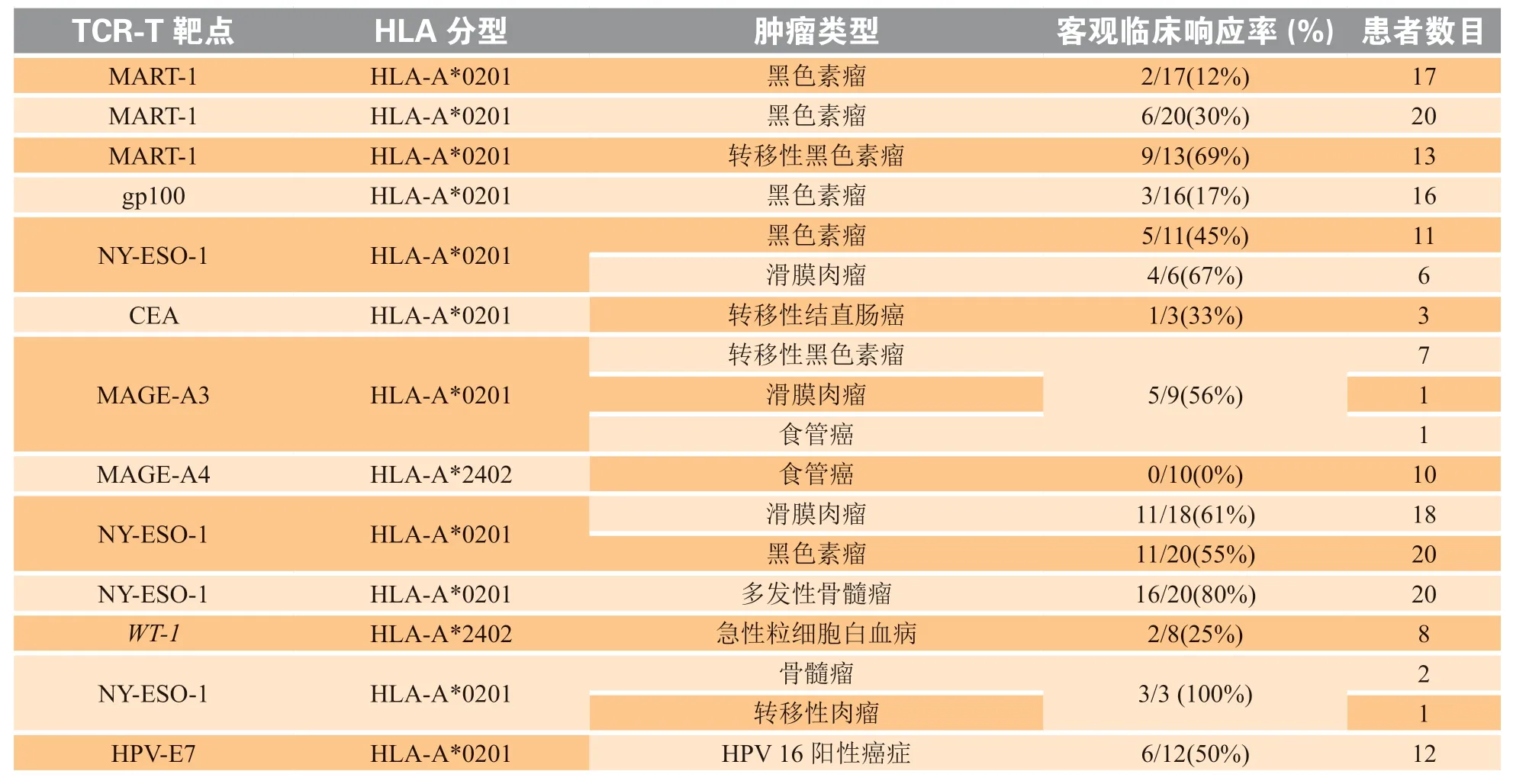
表1 已完成的T细胞受体工程化T细胞疗法临床试验结果Table 1 Results of conducted clinical trials of T cell receptor-engineered T cells therapy
4 动物模型在克服T细胞受体工程化T细胞 疗法挑战中的应用
尽管已有部分肿瘤患者从TCR-T疗法中获益[34],但作为一种新的肿瘤治疗方式,TCR-T疗法仍然具有一定的局限性。其限制性因素主要包括:靶向抗原的选择、肿瘤特异性TCR的开发、T细胞的体内持久性和TCR-T疗法的临床安全性等。为了更加安全有效地对肿瘤患者进行治疗,可以通过以下策略在非临床研究的动物模型中预测改善后的TCR-T疗法的抗肿瘤功效和临床安全性。
4.1 选择T细胞受体工程化T细胞靶向抗原
在TCR-T治疗过程中,第一步是选择最合适的肿瘤抗原作为TCR靶标。目前,主要存在3种类型的肿瘤抗原:存在于健康组织但在癌细胞中高表达的肿瘤相关抗原(tumor-associated antigens,TAAs);在癌细胞、睾丸、胎儿卵巢和滋养层细胞中表达的癌睾丸抗原[35](cancer testis antigens,CTAs);仅在癌细胞中表达的肿瘤特异性抗原(tumor-specific antigens,TSAs)如新抗原(neoantigen)和致癌病毒产生的抗原,但在健康组织中不表达。
目前,已有部分理想的肿瘤抗原短肽被发现,以用于TCR-T技术的开发。在这些抗原肽中,NYESO-1多次被证实是安全有效的肿瘤抗原之一,靶向该种抗原的TCR-T临床试验占30%以上。人乳头瘤病毒HPV-16中的E6和E7蛋白是肿瘤特异性抗原,与宫颈癌的发生密切相关,针对这2种抗原的TCR-T临床试验在这几年也迅速增加。此外,许多TAAs,CTAs或TSAs可用于TCR-T治疗,例如MART-1,威尔姆斯瘤基因1(Wilms' tumor gene 1,WT-1),乙型肝炎表面抗原(hepatitis B surface antigen,HBsAg),甲胎蛋白(alpha-fetoprotein,AFP),MAGE(A3,A4,A6,A10),黑色素瘤优先表达抗原(preferentially expressed antigen in melanoma,PRAME),巨细胞病毒(cytomegalovirus,CMV),EBV,糖胺聚糖(glycosaminoglycan,gag),gp100,红血球凝聚素1(hemagglutinin-1,HA1),人内源性逆转录病毒E(human endogenous retroviruses-E,HERV-E),大型多功能肽酶2(large multifunctional peptidase 2,LMP2),CEA,p53等[1],靶向这些抗原的临床试验也正在进行(见图1)。肿瘤新抗原由点突变、染色体易位和其他类型的致癌基因改变产生,这些抗原在除了肿瘤细胞外任何其他健康组织中均不存在,可作为TCR-T的靶点,而无脱靶毒性[36]。基因组测序、质谱分析技术和数据分析技术的进步促进了个体癌症特有的特异性新抗原的鉴定,使得自体TCR-T能够对具有临床活性的新抗原产生反应[37-39]。尽管有许多新抗原是通过经典突变产生的,但其他一些由翻译后修饰(例如磷酸化或甲基化)产生的新抗原也可以被TCR-T靶向[40-41]。

图1 T细胞受体工程化T细胞疗法的临床试验靶向抗原分布Figure 1 Distribution of targeted antigens of TCR-T clinical therapy
为了确保TCR-T疗法的安全性和有效性,原则上应该选择仅在癌细胞中表达的肿瘤特异性抗原作为理想靶标。筛选出的肿瘤抗原短肽在进入临床应用前需要在动物模型中验证其肿瘤特异性,证实这些抗原短肽是否仅在肿瘤细胞中表达,最大程度地减小TCR-T疗法在临床治疗中潜在的脱靶毒性。
4.2 肿瘤特异性T细胞受体的开发与优化
找到合适的肿瘤特异性抗原只解决了问题的一半,因为要筛选出靶向抗原的最适TCR也存在一定的挑战性。分离出肿瘤反应性T细胞需要选择合适的细胞来源,并且理想的T细胞要具有合适亲和力的TCR和显著的TCR表达频率与纯度,这些先决条件是成功开发出肿瘤特异性TCR的基础。获得肿瘤特异性T细胞后,在TCR测序中面临的最大挑战是每个T细胞肿瘤特异性TCR的α和β链的正确配对。高通量测序技术的引入可以使研究人员分析样品中数百万个TCR分子的多样性,通过这种方法可以获得所有TCR序列以构成样本库[42-44]。目前,获得特异性TCR最快的方法是将单细胞TCR测序与带有特定人类白细胞抗原(human leukocyte antigen,HLA)分子和选定肿瘤表位的条形码样多聚体结合起来,从而显著加快从人类样品中分离TCR的速度,避免任何富集步骤,并直接能够进行TCR序列的功能验证[45]。
TCR的亲和力被证实与TCR-T功能直接相关[46-48]。高亲和力的TCR已被用于大多数临床试验,因为它们能够以较低的表达水平识别肿瘤细胞,但是高亲和力的TCR可能会导致自身免疫性疾病。一些研究表明,TCR的亲和力处于低水平或中等水平也可以介导T细胞对肿瘤的杀伤,并且不会诱发自身免疫性疾病[49-53]。Miller等[49]研究发现,分别将高亲和力或低亲和力卵清蛋白(ovalbumin,OVA)特异性TCR-T转移至卵巢癌小鼠模型后,高亲和力的TCR-T在快速杀伤表达OVA的卵巢癌细胞的同时,也造成了小鼠的自身免疫性糖尿病;然而,低亲和力的TCR-T同样能介导肿瘤细胞的清除,而没有产生自身免疫性疾病。目前,关于中低亲和力TCR-T的良好抗肿瘤功效,只在部分肿瘤中得以体现,比如卵巢癌[49]、黑色素瘤[50-51]等,仍需更多的证据来证明中低亲和力TCR应用的普遍性。此外,有研究发现,某些化学药品、细胞因子和放射疗法可以激活HLA信号通路并上调肿瘤细胞表面抗原肽和HLA复合物的表达[54-55]。因此,使用具有最佳亲和力的TCR可以特异性地消灭肿瘤细胞,而不会诱发自身免疫性疾病,并且TCR-T疗法与其他疗法联合使用的组合治疗方案有望实现治疗效果最大化。
4.3 改善T细胞体内持久性
目前,工程化改造后的T细胞存在着体内持久性受限的问题。无论是传统的免疫细胞疗法,还是TCR-T疗法及CAR-T疗法,T细胞都需要在体外经刺激活化,大量增殖分化成为终末分化效应T细胞(effector T cell)后输回体内。回输的T细胞无增殖分化能力,且长期的体外刺激活化,使T细胞逐渐进入耗竭状态(T cell exhaustion),回输体内后容易走向死亡,而难以在体内发挥持久的抗肿瘤效应[56]。研究显示,记忆T细胞是一群低分化状态的异质性T细胞亚群,在持续对抗肿瘤或遇到复发肿瘤时能快速增殖分化产生效应T细胞而有效清除肿瘤细胞,在肿瘤免疫应答和免疫记忆维持中发挥重要作用[57]。
记忆T细胞在不同强度的信号刺激下,可根据表型、功能和转录本数据归为干细胞记忆T细胞(stem cell memory T cell,TSCM)、中枢记忆T细胞(central memory T cell,TCM)、效应记忆T细胞(effector memory T cell,TEM)及组织驻留记忆T细胞(tissue resident memory T cell,TRM)亚群[58]。Gattinoni等[59]发现,naïve T细胞激活分化后产生的TSCM仍具有naïve T细胞的表型,如CD45RA,CD62L及CCR7,又有激活后T细胞的表型,如CD95,IL2Rβ和IL15Rβ。作为记忆细胞,TSCM能快速地分泌细胞因子,响应IL-15快速地增殖,是一群自我更新能力最强的记忆T细胞亚群。Xu等[60]研究表明,在有IL-15和IL-7条件下体外培养T细胞时,能获得更高比例TSCM亚群,在小鼠肿瘤模型中的存活持续时间和抗肿瘤效果更好。TCM表达CD62L和CCR7,具有快速增殖和分化的特征,能够归巢至次级淋巴器官。TEM表型为CD62L-CCR7-,更倾向于分泌细胞毒因子,具有较强的溶瘤功能,可迁移至炎症组织中迅速发挥效应功能。TRM为组织驻留性记忆T细胞,不参与循环,由于其浸润能力以及直接的杀伤效应,能快速响应局部组织抗感染和抗肿瘤,在抗实体瘤中有应用前景[61]。
除了在临床前小鼠模型中,低分化水平的记忆T细胞在临床上也展现出更好的治疗效果。2019年,Chapuis等[62]报道了靶向抗原WT-1的TCR-T疗法的临床试验结果,对于TCR-T疗法响应较好的患者体内,低分化水平的中枢记忆T细胞所占的比例更高。因此,可以通过改善细胞培养条件或工程化改造T细胞等手段,使回输的TCR-T中具有更高比例的记忆性T细胞,从而提高T细胞回输后在体内的持久性,进而增强TCR-T疗法的抗肿瘤功效。
4.4 提高T细胞受体工程化T细胞疗法临床安全性
动物模型在提高TCR-T疗法的临床安全性方面起着关键作用。然而,在TCR-T疗法开发的过程中,这些动物模型也会出现无法准确预测细胞治疗毒性和移植物抗宿主反应等问题。Rapoport等[63]在以癌睾丸抗原NY-ESO-1为靶标对多发性骨髓瘤患者进行治疗的临床结果显示,虽然70%的患者病情接近完全缓解,但有5例患者出现了自体移植物抗宿主反应。Morgan等[64]在以黑色素瘤相关抗原MAGE-A3作为靶标的TCR-T临床试验结果显示,虽然部分患者出现缓解,但有3例患者出现了由脑部损害引起的精神障碍,其中2例受试者甚至面临死亡的危险,这是由于MAGE-A3 TCR交叉识别结合脑部正常组织中MAGE-A12抗原肽而导致的神经毒性。因此,不同肿瘤类型在选择特异性TCR时就意味着可能存在特定器官的毒性,如MAGE3 TCR在大脑和心脏中具有毒性。相较于CAR-T,小鼠模型在预测TCR的脱靶或靶标毒性方面有所欠缺。因此,用于TCR-T疗法研究的临床前小鼠模型还需进一步的改进与优化。在构建肿瘤模型时,选择表达了人细胞因子的人源化小鼠品系MITRG和MISTRG,为移植的人源细胞提供物种特异性细胞因子支持,可以更好地验证TCR-T疗法的治疗效果和临床安全性[65]。在TCR-T疗法的临床前研究中,选择构建人源肿瘤异种移植(patient-derived tumor xenograft,PDX)小鼠模型,将肿瘤患者的肿瘤组织移植至重症免疫缺陷型小鼠体内,使小鼠模型能更真实地模拟患者的体内环境,可为TCR-T治疗提供更精准的研究与评价工具[66]。此外,为了真实反映与评价人源免疫细胞与肿瘤微环境在抗肿瘤免疫治疗中发挥的作用,需要建立更加有效的人免疫反应体系小鼠模型。由于TCR-T发挥抗肿瘤作用离不开相关免疫细胞的参与,因此,构建含人源免疫细胞系统的人源化小鼠模型,在有效验证与评估抗肿瘤免疫治疗效果中可发挥至关重要的作用。
除了在非临床研究的动物模型中验证TCR-T疗法的临床安全性之外,还可以通过对TCR-T进行改造来增强TCR-T治疗的安全性。TCR-T疗法在临床治疗中往往展现出较CAR-T疗法更低的副作用,但脱靶效应造成的副作用仍阻碍着TCR-T疗法的发展。TCR-T疗法对肿瘤患者进行治疗的过程中,患者可能会出现CRS等严重的副作用,并可能进一步引起致命的并发症。若肿瘤抗原在健康组织细胞中低表达,则可能会引起TCR-T的靶点毒性,导致健康细胞被T细胞杀伤[67]。此外,外源TCR和T细胞内源TCR的错配也可能会导致TCR-T的非特异性脱靶毒性,而对正常组织造成严重损害[68]。此时,自杀基因的引入可作为调节和优化TCR-T疗法的备选策略。单纯疱疹病毒胸苷激酶(HSV-tk)是在临床上经过反复验证的一种自杀基因,并被证明安全有效,可以赋予TCR-T对更昔洛韦(ganciclovir)的致死敏感性[69-70]。其他自杀基因,比如截短的人类表皮生长因子受体(tEGFR)和诱导型Caspase 9安全开关(iCasp9)也经常被用于提高临床安全性的设计[71-72]。另外,临床前需要严格评估肿瘤靶向抗原是否会在正常组织或器官中表达。比如,在AFP特异性TCR-T疗法开展前,可利用丙氨酸扫描评估TCR-T的特异性和安全性[73]。由于外源TCR在T细胞中表达时,可能存在与内源TCR表达竞争和错配等问题,可以利用基因编辑和RNA干扰等技术,敲除或敲低内源性TCR,来避免这些问题[74-75]。
5 总结与展望
TCR-T免疫疗法正在逐渐发展成为一种广泛适用,且功能强大的癌症治疗手段。到目前为止,TCR-T疗法已在部分实体瘤的治疗中取得了较好的临床效果,特别是针对黑色素瘤和滑膜细胞肉瘤等的治疗。尽管TCR-T疗法为肿瘤患者带来了新的治疗选择和希望,但是该疗法对于实体瘤的治疗还存在一些挑战,包括靶向抗原的选择、肿瘤特异性TCR的开发、T细胞的体内持久性和TCR-T疗法的临床安全性等。相信随着工程化改造T细胞研究的不断发展,在不久的将来会有更多的肿瘤患者在TCR-T疗法中受益。
