催化碳烟氧化用PtCexZr1-xO2催化剂的制备及性能研究
2021-11-06朱敬芳杜奥林高琨阳常仕英赵云昆杨冬霞潘再富顾永万
朱敬芳,杜奥林,高琨阳,常仕英,赵云昆,杨冬霞,潘再富,顾永万*
(1. 昆明贵研催化剂有限责任公司,昆明 650106;2. 昆明贵金属研究所 稀贵金属综合利用新技术国家重点实验室,昆明 650106)
柴油发动机因其高效性、经济性、耐久性和低碳排放等特点,在商业上受到越来越多的关注[1],但柴油发动机尾气排放中会产生大量颗粒物(PM)和氮氧化物(NOx),严重危害环境和人体健康[2]。因此国家针对柴油车尾气排放限值的法规要求越来越严格,尤其是GB17691-2018《重型柴油车污染物排放限值及测量方法(中国第六阶段)》的实施给柴油车尾气后处理系统带来了巨大的挑战。PM的净化主要通过在柴油车后处理系统中安装颗粒物捕集器(DPF)将其收集,然后在合适的条件下进行再生处理,壁流式的DPF是目前收集PM最常用的一种方式[3-4]。碳烟是PM的主要成分[5],在没有催化剂作用下,碳烟要在450℃以上才开始氧化,但柴油机的排气温度一般在200℃~500℃之间,为了使DPF捕集的碳烟能够迅速再生,满足法规对碳烟的排放控制要求,通常采用的方式有两种,一种是通过提高尾气或DPF本身的温度来使碳烟氧化,另一种是通过涂覆催化材料到壁流式的DPF陶瓷载体上制备得到CDPF催化剂来降低碳烟的氧化温度[6]。因此开发具有低温氧化活性的碳烟催化材料,降低碳烟氧化温度,加快氧化速率,降低再生频率,延长CDPF催化剂的寿命,对柴油车尾气后处理系统具有重要意义。
由于CeO2具有优异的储氧能力,被广泛应用在汽车尾气净化催化剂中[7]。Setiabudi等人研究了CeO2在催化碳烟氧化中的作用,发现其对碳烟的氧化性能与活性氧储存性能有关[8];CeO2与其他金属氧化物复合而成的催化剂在催化碳烟氧化上也具有很好的活性,受到大量关注[9-10],在CeO2中引入ZrO2可以增强催化剂的热稳定性和储氧性能,进一步提升催化碳烟氧化的活性[11-12]。柴油车排放的尾气中含有大量的NOx,其主要成分是NO,贵金属铂(Pt)对NO氧化具有优异的催化活性[13],能够将NO氧化为NO2,而NO2的氧化性能优于O2,更有利于碳烟的氧化,因此Pt作为活性金属用于碳烟氧化催化剂的研究较多[14-19]。目前催化碳烟氧化催化剂的制备方法主要是采用传统的浸渍法,将Pt负载在CeZrOx复合氧化物材料上获得Pt/CexZr1-xO2催化剂;采用水热晶化法一步法[20]将贵金属前驱体和基础CeZrOx复合氧化物混合一起制备的研究较少见。
本研究通过将铂(Pt)的前驱体与CeO2、ZrO2的前驱体混合,采用水热晶化法一步制备PtCexZr1-xO2系列催化剂,研究铈锆摩尔比(nCe/nZr)对系列催化剂的结构、物化性能以及催化碳烟氧化活性的影响;并将该一步法制备的PtCexZr1-xO2催化剂与传统方法制备的Pt/CexZr1-xO2催化剂在结构、物化性能、催化碳烟氧化性能进行比较,为碳烟氧化催化剂的开发、设计提供应用指导。
1 实验
1.1 材料及设备
六水合硝酸铈(Ce(NO3)3·6H2O),五水合硝酸锆(Zr(NO3)4·5H2O),聚乙二醇(PEG-6000),氨水(浓度:25%),无水乙醇,上述试剂均为分析纯,购自国药试剂;氯铂酸溶液(铂质量分数为15%)由贵研铂业有限责任公司提供;实验用水为去离子水。
制备用水热反应釜内衬罐为聚四氟乙烯材质,容积150 mL,最高工作温度200℃。
1.2 水热晶化法制备PtCexZr1-xO2系列催化剂
1) 新鲜催化剂的制备。采用水热晶化法一步制备铈锆摩尔比不同的PtCexZr1-xO2系列催化剂各20 g,其中nCe/nZr分别为1:9,3:7,5:5,7:3,9:1,样品的编号根据复合物中铈锆的摩尔比进行定义(见后文表1)。根据不同的铈锆摩尔比,称取对应量的Ce(NO3)3·6H2O和Zr(NO3)4·5H2O,放入40℃的去离子水中,使其充分溶解,冷却至室温。加入铂浓度(质量分数)为1%的氯铂酸溶液,最后加入表面活性剂PEG-6000,加入量为铈锆复合物摩尔量的30%,持续搅拌20 min,使其溶解,滴加氨水进行沉淀,当pH=9时停止沉淀。将沉淀倒入水热晶化反应釜中,在120℃下连续水热晶化12 h。对晶化后的沉淀进行离心洗涤,先用去离子水洗涤3次,再用无水乙醇洗涤1次,将最终获取的沉淀放入坩埚内,将坩埚放入鼓风干燥箱内,于120℃干燥24 h,将坩埚转入马弗炉内,在空气气氛中,600℃焙烧3 h,制备得到催化剂新鲜(Fresh)样品,编号以尾注“-F”标识。
2) 老化催化剂的制备。将催化剂新鲜样品放入马弗炉中,在空气气氛中,800℃焙烧4 h,制备得到催化剂老化(Aged)样品,编号以尾注“-A”标识。
1.3 浸渍法制备Pt/Ce0.5Zr0.5O2催化剂
参考此前的制备方法[20],以商用Ce0.5Zr0.5O2材料为载体,采用浸渍法制备铂负载量为1%的Pt/Ce0.5Zr0.5O2催化剂。量取准确量的氯铂酸溶液(所含Pt质量折算为载体质量的1%),用20 mL去离子水稀释;将Ce0.5Zr0.5O2材料浸渍到稀释后的氯铂酸溶液中,室温下搅拌2 h,静置2 h。将浸渍后的溶液放置于鼓风干燥箱中,120℃干燥24 h。干燥后得到的粉末放入坩埚中,将坩埚转移到马弗炉中,在空气气氛中,600℃焙烧3 h。制备得到的催化剂新鲜样品标记为Pt/Ce0.5Zr0.5O2-F;将催化剂新鲜样品放入马弗炉中,在空气气氛中,800℃焙烧4 h,得到催化剂老化样品,标记为Pt/Ce0.5Zr0.5O2-A。
1.4 催化剂的物化性能表征
催化剂的X射线衍射(XRD)表征、比表面积(SBET)测定、储氧量(OSC)测定、还原温度测试所用方法与前文[20]相同。
1.5 催化活性测试
催化活性在一套具备多管路的连续微型固定床反应器中进行[20]。将催化剂样品与碳烟按照质量比为20:1的比例以紧密接触方式混合(玛瑙研钵中研磨10 min),经过压片制作样品,称量样品0.5 g,用于催化剂活性评价。测试气体组成(体积分数)为NO 400×10-6、O28%,H2O 3%,N2作为平衡气,气体空速为40000 h-1;程序升温从100℃升温至600℃,600℃保温10 min。用傅里叶红外光谱分析仪(美国MKS 2030)检测不同温度下反应器后端CO和CO2的浓度,评价催化剂的活性。
2 结果与讨论
2.1 催化剂的晶相结构
图1为催化剂的XRD谱图,表1为用Scherrer公式计算得到的催化剂晶粒尺寸数据。

表1 催化剂的晶粒尺寸Tab.1 Crystallite size data of catalysts
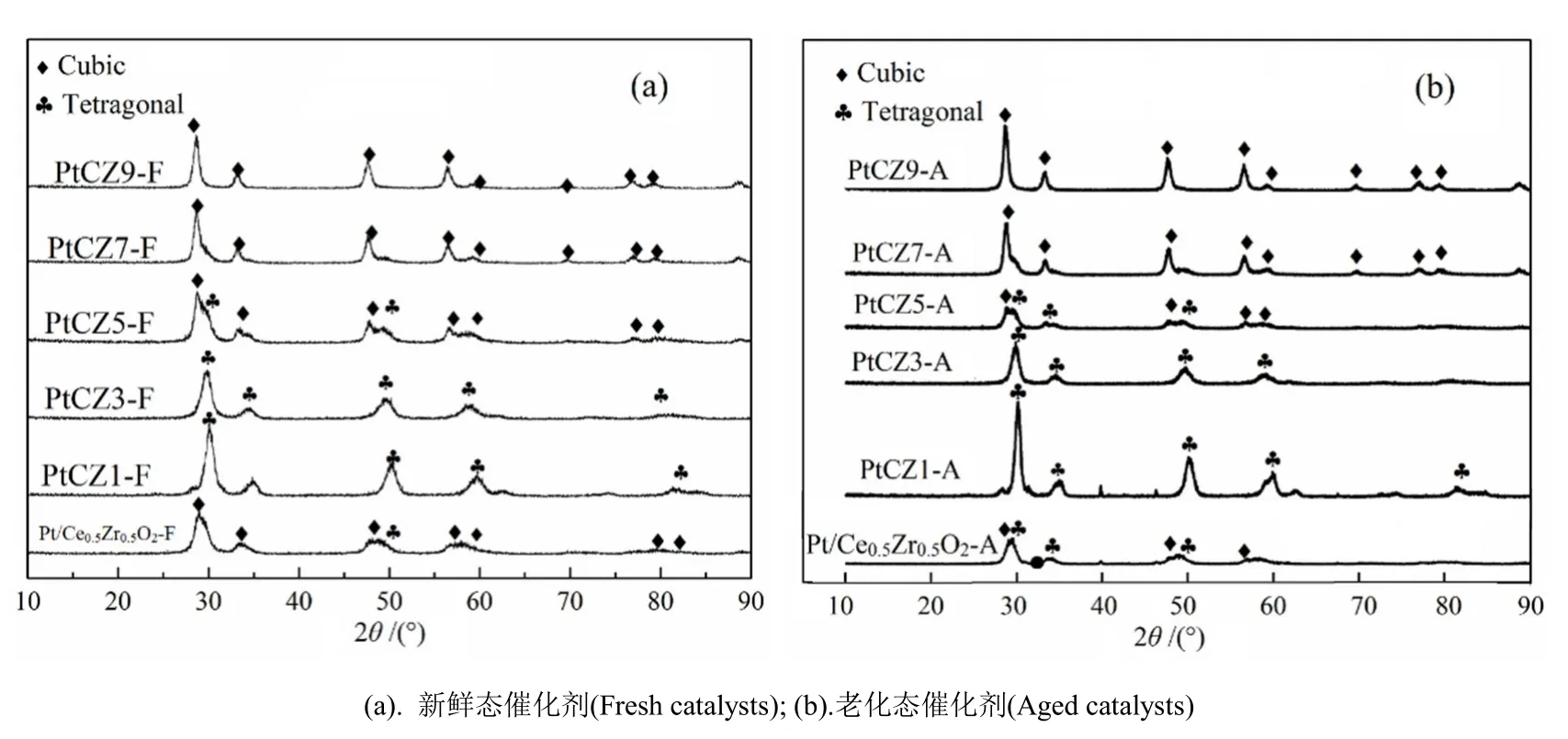
图1 催化剂的XRD谱图Fig.1 The XRD patterns of catalysts
图1(a)表明,所有催化剂的XRD谱图中均没有铂的特征峰出现,均为铈锆复合物的特征峰。所有催化剂的特征峰都较立方相CeO2的特征峰(标准卡片JCPDS 65-5923)向高角度有所偏移,这主要是由于Zr4+的引入导致CeO2的晶格收缩造成的[21]。当nCe/nZr=9:1时,催化剂PtCZ9-F的2θ特征峰分别出现在28.74°、33.08°、47.6°、56.42°、59.38°、69.5°、76.88°、79.33°和88.76°,均归属为立方相结构。因为催化剂PtCZ9-F组分中ZrO2含量较少,其特征峰位置相对于立方相CeO2的特征峰略微向高角度偏移。随着催化剂组分中ZrO2含量的增加,催化剂特征峰的位置向高角度偏移更多,且催化剂的晶相不再单纯为立方单相。当nCe/nZr=5:5,催化剂PtCZ5-F的特征峰位置偏移量明显变大,并在2θ为34.54°和49.64°处出现了明显的四方相铈锆固溶体的特征峰(标准卡片JCPDS 38-1436),说明催化剂PtCZ5-F为立方相和四方相2种晶相共存的固溶体。当nCe/nZr= 3:7时,催化剂PtCZ3-F的特征峰位置向高温移动,且特征峰归属为四方相铈锆固溶体,说明催化剂PtCZ3-F为四方相结构。当nCe/nZr= 1:9时,催化剂PtCZ1-F的2θ特征峰分别出现在30.2°、34.36°、34.86°、50.04°、59.278°、62.52°、74.28°、81.38°和84.22°,这与四方相ZrO2的特征峰(标准卡片JCPDS 79-1765)非常接近,说明催化剂PtCZ1-F为四方相结构。上述结果与文献[22]报道一致,当nCe/nZr≥5:5时,铈锆复合物主要以立方晶相存在,小于5:5时主要以四方晶相存在。用浸渍法制备的Pt/Ce0.5Zr0.5O2-F催化剂的XRD结果表明,谱图中并未出现铂的衍射峰,均是Ce0.5Zr0.5O2复合氧化物的特征峰,与PtCZ5-F催化剂的出峰位置一致,为立方相和四方相共存的固溶体。主要原因可能是因为本研究中铂的含量低,少量添加铂并未影响铈锆复合氧化物的晶相。
从图1(b)可以看出,催化剂经过高温焙烧老化后,特征峰的位置没有发生变化,说明高温焙烧老化过程没有改变催化剂的晶相结构。老化后,大部分催化剂特征峰的衍射强度都明显增强,峰形变得更加尖锐,这主要是由于样品在老化的过程中发生了不同程度的烧结及团聚造成的[23]。但PtCZ5-A和Pt/Ce0.5Zr0.5O2-A两个催化剂的特征峰形较新鲜催化剂的特征峰形却没有变尖锐,说明在老化的过程中,催化剂的结构得到较好的保持,烧结造成的团聚现象不明显。
表1数据显示,具有立方相和四方相晶格结构的催化剂PtCZ5-F晶粒尺寸最小,具有立方相晶格结构的催化剂PtCZ9-F,PtCZ7-F的晶粒尺寸较具有四方相晶格的催化剂PtCZ3-F,PtCZ1-F的晶粒尺寸大,结果与文献[24]报道的铈锆复合物的晶粒尺寸与铈锆摩尔比的规律一致,说明PtCeZrOx催化剂的中铂的加入没有改变铈锆复合物的晶粒尺寸大小。经过高温老化后,催化剂的晶粒尺寸均有所增加,是由于催化剂在老化过程中团聚造成的[23]。浸渍法制备的催化剂Pt/Ce0.5Zr0.5O2在新鲜态和老化态时,其晶格尺寸均较同样铈锆比例的催化剂PtCZ5-F的晶粒尺寸略大。
2.2 催化剂的比表面积和储氧量
通过N2-物理吸附表征,获取催化剂的比表面积,结果如表2所示。

表2 催化剂的比表面积和储氧量Tab.2 BET and oxygen storage capacities of catalysts
表2中比表面积数据显示,PtCZ1-F、PtCZ3-F和PtCZ5-F三种催化剂新鲜态时比表面积较大,而PtCZ7-F、PtCZ9-F比表面积较小。主要是由于催化剂组成中ZrO2和CeO2的含量不同造成的。ZrO2的加入有利于增加铈锆复合物的比表面积[3-4],因此当催化剂组成中ZrO2的含量高时,催化剂的比表面积较大。经过高温焙烧老化后,催化剂的比表面积都有所衰减,主要是由于在高温焙烧的过程中催化剂的内部孔道结构坍塌造成比表面积的减少[23]。浸渍法制备的催化剂Pt/Ce0.5Zr0.5O2其比表面积在新鲜态和老化态下均小于催化剂PtCZ5,说明水热晶化法较浸渍法制备的催化剂比表面积更大,更加有利于碳烟与催化剂的接触。
表2中储氧量(OSC)结果表明,PtCZ3-F、PtCZ5-F、PtCZ7-F三种催化剂的储氧量较PtCZ1-F、PtCZ9-F高,这与这与文献[25]的研究结果相吻合,当铈锆固溶体中铈的含量为20%~50%时,相对其它比例的铈锆固溶体拥有更高的总储氧量。因为催化剂的储氧量主要取决于铈锆复合物的储氧量,而铈锆复合物的储氧量主要是CeO2中的氧空穴形成,加入ZrO2后,由于Zr4+嵌入到CeO2的晶格中,导致CeO2的晶格收缩,形成更多的氧空穴,催化剂的储氧量得到提升[26]。经过高温焙烧老化后,所有催化剂的储氧量较新鲜催化剂均降低,主要是高温焙烧过程中孔道结构的坍塌造成的。传统浸渍法制备的Pt/Ce0.5Zr0.5O2催化剂的储氧量新鲜态与催化剂PtCZ5一致,但老化后其储氧量小于催化剂PtCZ5,其比表面积也低于催化剂PtCZ5,说明高温焙烧的老化过程对催化剂Pt/Ce0.5Zr0.5O2的结构坍塌、表面团聚更加明显。
2.3 催化剂的氢气还原性能
图2为催化剂的H2-TPR谱图。根据文献[27]报道铈锆复合物中铈锆的含量对铈锆复合物的表面氧和体相氧的出峰位置有影响,其中铈锆复合氧化物表面氧的还原峰出峰位置较体相氧的出峰温度更低,在500℃附近。
图2结果表明,本研究中催化剂的还原峰位置主要在200℃~400℃之间,较铈锆复合物表面氧的还原峰出峰位置向低温偏移,主要是由于催化剂PtCexZr1-xO2较纯的铈锆复合物增加了活性组分Pt,Pt加强了表面氧的传输速率,因此表面氧的出峰位置向低温移动,催化剂还原峰在250℃~400℃之间。
图2(a)表明,催化剂PtCZ1-F和PtCZ9-F的还原峰温度更低,说明两种催化剂能在更低的温度下被氢气还原,但两种催化剂的还原峰面积较小,说明两种催化剂的表面氧含量较低,尤其是PtCZ1-F的还原峰面积最小,主要是由于PtCZ1-F的组分中CeO2的含量比较低,储氧量低,因此表面氧含量低。催化剂PtCZ5-F的还原温度、峰面积均优于催化剂PtCZ7-F、PtCZ3-F和Pt/Ce0.5Zr0.5O2,说明催化剂PtCZ5-F具有优异的氢气还原温度性能和较高的表面氧含量。

图2 催化剂的H2-TPR谱图Fig.2 H2-TPR profiles of catalysts
图2(b)结果表明,催化剂经过高温老化后,还原峰的位置向高温移动且峰面积有所减小,主要是由于老化过程中高温煅烧导致催化剂内部孔道坍塌、团聚造成的比表面积减小、储氧量降低造成的。
2.4 催化剂的催化活性
本研究的反应过程中主要发生了以下反应:
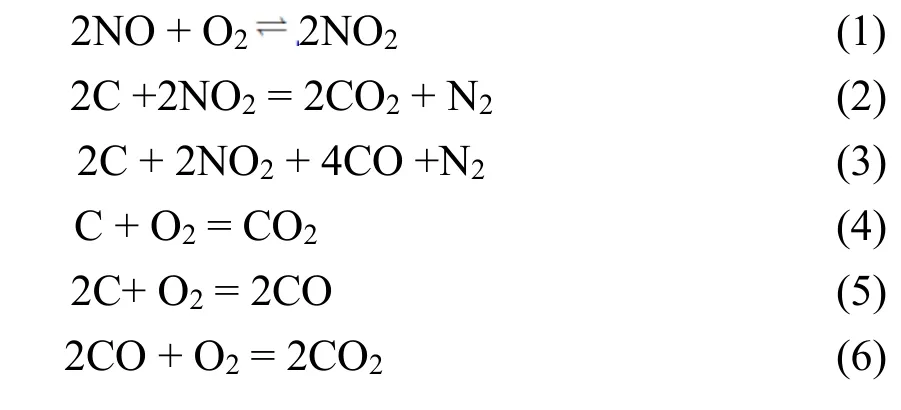
催化剂的活性主要通过测试碳烟氧化后生成的CO2的温度进行评价,使用Ti(碳烟开始氧化的温度)、Tm(碳烟氧化过程中生成的CO2浓度最大时对应的温度)、Tf(碳烟氧化完的温度)和ΔT(ΔT=Tm-Ti,反映碳烟氧化速度)作为催化剂性能的评价参数。其中Ti和Tm越低说明催化剂的低温氧化活性越好,能够在较低的温度下催化碳烟氧化,ΔT越小说明碳烟的氧化速率越快,催化剂的活性越好[28]。
碳烟氧化的产物主要是CO2,若碳烟未完全反应会有副产物CO的生成,在PtCexZr1-xO2系列催化剂作用下,碳烟氧化后未检测到CO成分,说明PtCexZr1-xO2不仅能够催化碳烟在低温下氧化,还能够促进碳烟氧化完全生成CO2,不再产生新的污染物CO。PtCexZr1-xO2催化碳烟氧化产生的CO2浓度曲线如图3所示,得到的各特征温度数据列于表3。

表3 催化剂PtCexZr1-xO2系列催化碳烟氧化的特征温度Tab.3 The temperatures of soot oxidation on PtCexZr1-xO2 catalysts with various cerium-zirconium ratios /℃
图3(a)曲线表明,水热晶化法新制备的PtCexZr1-xO2催化碳烟氧化活性的排序为PtCZ5-F>PtCZ9-F>PtCZ7-F>PtCZ3-F>PtCZ1-F,其中PtCZ5-F能在较低的温度下氧化碳烟,催化氧化性能最优。
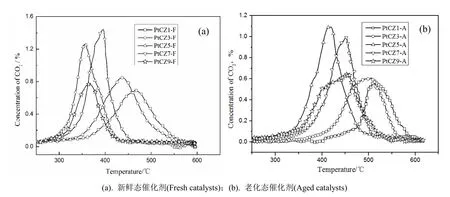
图3 PtCexZr1-xO2催化碳烟氧化生成的CO2的浓度曲线图Fig.3 The concentrations of CO2 by soot oxidation on PtCexZr1-xO2 catalysts
根据表3数据,PtCZ5-F对碳烟氧化的Ti、Tm和Tf分别为283℃、356℃和430℃,较其他催化剂作用下碳烟的氧化温度低;且ΔT=73℃也是所有催化剂中最小的。说明PtCZ5-F对碳烟的氧化速率快,对柴油车后处理中CDPF催化剂中碳烟再生非常有利。碳烟不仅能够在较低的温度下开始氧化,并且能够快速完成再生,缩短再生时间,提高CDPF催化剂的再生效率。结合前文数据,根据XRD结果,催化剂PtCZ5-F为四方相和立方相共存结果,晶粒尺寸最小;储氧量和H2-TPR的结果显示,催化剂PtCZ5-F的OSC值虽然不是最高,但H2-TPR结果表明其表面氧含量最高,H2还原温度较低;BET的数据显示,催化剂PtCZ5-F的比表面积较大,综合催化剂的催化活性和催化剂的结构表征、物化性质表征数据,说明在催化剂PtCexZr1-xO2系列中,催化剂的表面氧含量、H2还原温度、晶粒尺寸大小对催化碳烟氧化的活性影响较大。
催化剂老化后催化碳烟氧化的温度较新鲜状态时升高,主要是由于催化剂在高温老化的过程中,内部结构孔道的坍塌和晶粒尺寸变大、表面氧含量降低引起的。催化剂PtCexZr1-xO2系列对碳烟氧化的活性排序跟新鲜态一致,催化剂PtCZ5-A对碳烟氧化的催化活性最优。
2.5 不同方法制备催化剂性能对比
图4为传统浸渍法制备的催化剂Pt/Ce0.5Zr0.5O2与水热晶化法制备的活性最优的催化剂PtCZ5催化碳烟氧化活性曲线,表4列出了特征温度对比。
图4和表4结果表明,水热晶化法制备的催化剂PtCZ5在新鲜态和老化态时,较传统浸渍法制备的催化剂Pt/Ce0.5Zr0.5O2对催化碳烟氧化的低温活性都更优。这主要是由于PtCZ5的表面氧含量、还原温度优于Pt/Ce0.5Zr0.5O2,其更容易释放出活性氧与碳烟接触,更利于催化碳烟低温氧化。

图4 两种催化剂氧化碳烟生成的CO2的浓度曲线Fig.4 The concentrations of CO2 by soot oxidation on the two catalysts

表4 两种催化剂催化碳烟氧化的特征温度Tab.4 The temperatures of soot oxidation on the two catalysts /℃
3 结论
1) 采用水热晶化法一步合成不同铈锆摩尔比(nCe/nZr)的催化剂PtCexZr1-xO2系列,其晶相结构、晶粒尺寸、比表面积、储氧性能、H2-还原温度、表面氧的含量不同,造成催化剂催化碳烟氧化的活性不同。其中晶相结构、晶粒尺寸、表面氧含量、还原温度对催化剂催化活性影响较大。
2)nCe/nZr= 5:5制备的催化剂PtCZ-5,新鲜态和老化态均含有四方相和立方相2种晶型结构,且晶粒尺寸最小,表面氧含量最高,催化碳烟氧化活性最强,碳烟开始燃烧温度为283℃。较其他锆铈摩尔比制备的催化剂、浸渍法制备的Pt/Ce0.5Zr0.5O2催化剂对碳烟氧化的催化温度更低。在柴油车尾气后处理系统中碳烟颗粒捕集催化剂(CDPF)进行碳烟再生(氧化)时,水热晶化法制备的PtCe0.5Zr0.5O2催化剂具有较好的应用前景。
