利用可诱导表达Cas9的HeLa细胞系进行顺铂耐药性基因筛选
2021-11-05陈红全王建瑛
陈红全,王建瑛
1浙江大学医学院附属邵逸夫医院检验科,浙江 杭州 310016;2南京医科大学生殖医学国家重点实验室,江苏 南京 211166
耐药性仍然是治愈癌症的主要限制因素,尽管多种化疗药物早期对肿瘤的治疗效果很好,但随着耐药性的产生,常导致癌症复发[1],对肿瘤细胞耐药机制的研究有助于更好地治疗癌症。RNA 干扰技术(RNA interference,RNAi)曾被应用于肿瘤细胞耐药基因的筛选,但由于RNAi 不能完全抑制基因表达,而且存在广泛的脱靶效应,影响对基因功能的判定。CRISPR⁃Cas9(clustered regularly interspaced short palindromic repeats/Cas9)系统具有高效、便捷等诸多优点,为肿瘤细胞耐药性分子机制的解析提供了强大的工具[2]。
CRISPR⁃Cas 系统是细菌和古细菌在长期演化过程中形成的免疫防御系统,用以对抗入侵的病毒及外源核酸[3]。目前,对Ⅱ类CRISPR⁃Cas系统中来源于酿脓链球菌的Streptococcus pyogenesCas9(sp⁃Cas9)研究最为深入,Cas9蛋白作为核酸内切酶,在引导RNA 的指导下识别目标DNA 序列后激活Cas9蛋白的酶切活性[4]。正是由于这种靶向功能,CRIS⁃PR/Cas9 系统已被广泛应用于多物种的基因编辑、基因调控、定位成像、细胞谱系追踪、高通量文库筛选以及病毒检测等领域[5]。目前该系统还存在诸多缺点,如Cas9表达载体过大,不容易被转染进细胞;Cas9载体长时间表达,会增加脱靶的风险[6]。因此,有必要构建一个可诱导表达Cas9的稳定细胞系,用以解决低效转染和因Cas9 长时间表达而导致高脱靶风险的问题。
慢病毒常被用于稳定细胞系的构建,但是需要包装病毒颗粒,费时费力,并存在感染的风险,本研究利用便捷的PiggyBac 转座子系统,成功构建了可诱导表达Cas9 的稳定HeLa 细胞系,并证明在多个位点可以高效切割基因组;同时,我们针对人核受体基因,设计了对应的sgRNA 文库,在这个细胞系上进行了抗癌药物顺铂(Cisplatin)耐药性的筛选,以期为顺铂耐药机制的解析打下基础。
1 材料和方法
1.1 材料
HEK293T 和HeLa 细胞购自美国模式培养物集存库(American Type Culture Collection,ATCC);高糖DMEM(HyClone公司,美国),胎牛血清(FBS)(Gem⁃ini 公司,美国),青链霉素(Gibco 公司,美国);Lipo⁃fectamine 2000(Invitrogen 公司,美国);强力霉素(Sigma 公司,美国);Zeocin(Invitrogen 公司,美国);嘌呤霉素(Merck 公司,德国);高保真酶PrimeSTAR Max DNA Polymerase(TaKaRa 公司,日本);限制性内切酶BsaⅠ(NEB 公司,美国);Esp3 Ⅰ(Fermentas公司,立陶宛);多聚赖氨酸(Sigma 公司,美国);T7EN1(NEB 公司,美国);T 载体(TaKaRa 公司,日本)。
1.2 方法
1.2.1 质粒构建
pST1374⁃NLS⁃Flag⁃Cas9、pST1374⁃NLS⁃Flag⁃Cas9⁃NLS、pST1374⁃NLS⁃Flag⁃Cas9⁃NP、pST1374⁃Cas9⁃NLS 和pST1374⁃Cas9⁃SP 改自pST1374⁃NLS⁃flag⁃linker⁃Cas9(Addgene 公司,美国)[7]。用于构建针对人RAG1基因的sgRNA表达载体通过寡聚核苷酸(oligo)在体外退火,利用BsaⅠ位点插入pGL3⁃U6⁃sgRNA⁃PGK⁃Puro 载体(Addgene 公司,美国)[7],所用oligo 序列为hRag1⁃U6⁃Up:5′⁃CCGGTGTCT⁃GCTTTGATGGACA⁃3′;hRag1⁃U6⁃Down:5′⁃AAACT⁃GTCCATCAAAGCAGACA⁃3′。
用于高通量构建sgRNA 的表达载体pGL3⁃U6⁃ccdB⁃PGK⁃Puro 改自pGL3⁃U6⁃sgRNA⁃PGK⁃Puro 载体。以pGL3⁃U6⁃sgRNA⁃PGK⁃Puro 为模板,利用前向特异引物和共用反向引物扩增出19N⁃sgRNA backbone,通过Esp3Ⅰ酶切位点,插入pGL3⁃U6⁃ccdB⁃PGK⁃Puro 载体。用于构建sgRNA 文库的慢病毒载体pLKO.1⁃U6⁃ccdB⁃PGK⁃Puro 改自pGL3⁃U6⁃ccdB⁃PGK⁃Puro。
针对47个核受体基因各设计2条sgRNA(表1)。共用的反向引物为T7 gRNA Esp3I Rev:5′⁃TTA⁃CACCGTCTCGTTTATGCTTCCGGCTCGTATG⁃3′。

表1 构建sgRNA表达载体所用引物Table 1 Sequences of primers for constructing sgRNA expression vectors
(续表1)
PiggyBac 转座子系统由刘澎涛教授惠赠[8]。NLS⁃Flag⁃Cas9⁃NLS 和Cas9⁃NLS 片段分别来自pST1374⁃NLS⁃Flag⁃Cas9⁃NLS 和pST1374⁃Cas9⁃NLS,IRES⁃hrGFP 片段扩增自pShuttle⁃IRES⁃hrGFP(Agi⁃lent Technologies 公司,美国),Sv40 Pro⁃BleoR⁃pA 片段扩增自pcDNA3.1⁃Zeo载体,以上片段通过酶切位点克隆至PiggyBac 表达载体中,构建PB⁃TRE⁃NLS⁃Flag⁃Cas9⁃NLS⁃IRES⁃hrGFP⁃Zeo 和PB⁃TRE⁃Cas9⁃NLS⁃IRES⁃hrGFP⁃Zeo载体。
1.2.2 细胞培养和转染
HEK293T 和HeLa 细胞培养于高糖DMEM,10%FBS,1%100×青链霉素中。
HEK293T 细胞利用Lipofectamine 2000 进行转染。在12孔板中,1 μg Cas9各载体和0.5 μg sgRNA载体被共转染到HEK293T中,72 h之后收集细胞用于下游分析。在24 孔板中,0.5 μg PB⁃TRE⁃NLS⁃Flag⁃Cas9⁃NLS⁃IRES⁃hrGFP⁃Zeo 和0.25 μg PB⁃CAG⁃rtTA载体被共转染到HEK293T中,24 h之后加入强力霉素(Dox)至2 μg/mL,72 h之后用于表达分析。
HeLa细胞利用电转(BioRad Gene Pulser XL)转染。4 μg PB⁃TRE⁃Cas9⁃NLS⁃IRES⁃hrGFP⁃Zeo、2 μg PB⁃CAG⁃rtTA和2 μg转座酶载体被共电转到2×106个HeLa细胞中,48 h之后,加入100 μg/mL Zeocin筛选3 代,然后用于单克隆的生长和挑选。对于挑出来的稳定表达可诱导Cas9 的细胞株,电转2 μg sgRNA 表达载体,24 h 后加入Dox(2 μg/mL)和嘌呤霉素(1 μg/mL),72 h后收集细胞并提取DNA。
HeLa8E 克隆细胞经扩增后加入Dox,以诱导Cas9 的表达和对目标基因的编辑。分别加入0.3、0.9、1.5 μg/mL 顺铂处理细胞48 h,在对照细胞(未加Dox)全部死亡之后,撤掉顺铂。由于0.3 μg/mL的顺铂不能使对照细胞全部死亡,后续实验从0.9 μg/mL和1.5 μg/mL两组中挑选单克隆进行。
1.2.3 免疫荧光染色
HEK293T 细胞培养在包被了多聚赖氨酸的盖玻片上,转染72 h之后,细胞直接用于拍照,或者用4%的PFA(paraformaldehyde)室温固定15 min,PBS洗2 次,用含0.2%Triton 100 的PBS 处理5 min 以增加细胞膜的渗透性,然后用标准羊血清(武汉博士德公司)室温封闭30 min。用加抗⁃Flag M2(Sigma公司,美国)的封闭液室温孵育2 h。PBS 洗2 次后,将cy3⁃conjugated⁃goat⁃抗⁃mouse IgG(Jackson Immuno⁃Research 公司,美国)二抗和Hoechst 33258(Sigma公司,美国)加入PBS,并室温孵育细胞1 h,PBS 洗3 次后用Vectashield Mounting medium(Vector Laborato⁃ries 公司,日本)进行封片。所有免疫荧光图片用Olympus Flueview 1000 激光共聚焦显微镜拍摄。
1.2.4 T7EN1切割检测
收集的细胞用裂解液(10 μmol/L Tris⁃HCl,0.4 mol/L NaCl,2 μmol/L EDTA,1% SDS,100 μg/mL Proteinase K)在58 ℃水浴锅中消化过夜,用酚氯仿抽提裂解物,然后用乙醇沉淀得到DNA。目的DNA片段通过PCR 从基因组中扩增出,PCR 产物用PCR cleanup kit(Axygen 公司,美国)纯化,吸取200 ng 纯化后的PCR 产物,在20 μL 的1×NEBuffer 2(NEB 公司,美国)体系中退火,退火程序为95 ℃5 min;95 ℃~85 ℃,每秒降低1 ℃;85 ℃~25 ℃,每秒降0.1 ℃;4 ℃保持。退火后的PCR 产物用0.5 μL T7EN1 在37 ℃水浴锅中处理30 min,然后在2.5%的琼脂糖凝胶中电泳。利用Image J 软件对凝胶电泳图片进行光密度定量分析,采用之前报道的公式计算切割效率[9],公式如下:切割效率=。a、b代表被切割片段的光密度值,c 代表未被切割片段的光密度值,对于需要TA 克隆和测序的样品,PCR产物克隆进T载体,挑取阳性克隆用M13⁃47通用引物测序。
PCR扩增引物hRAG1⁃For:5′⁃GTGAAAGCCAT⁃CACAGGGAGAC⁃3′,hRAG1⁃Rev:5′⁃AGAATGCCT⁃CCCAGCTCAAGC⁃3′,扩增产物大小为623 bp;hERb lib⁃testF:5′⁃CTTCCTCCTACAACTGCAGTCAA⁃3′,hERb lib⁃testR:5′⁃CGCAATCGTGCCATTATACT⁃CCA⁃3′,扩增产物大小为656 bp;hERRa lib⁃testF:5′⁃GCATTGAGCCTCTCTACATCAAG⁃3′,hERRa lib⁃tes⁃tR:5′⁃GGTACCCTGAACTCACTCACATC⁃3′,扩增产物大小为522 bp;hERRb lib⁃testF:5′⁃GACTTCCC⁃GATTTGTGTCCACAG⁃3′,hERRb lib⁃testR:5′⁃CT⁃GTCCAGTGCCAGAATGAGAAG⁃3′,扩增产物大小为609 bp;hERRg lib⁃testF:5′⁃CACTACAGGACAGAC⁃GCTTCATT⁃3′,hERRg lib⁃testR:5′⁃CTCGTTAGCT⁃TACACTGGCAGTA⁃3′,扩增产物大小为684 bp。
1.2.5 顺铂耐药克隆sgRNA 序列测定和目标基因验证
顺铂耐药克隆sgRNA 序列扩增引物为pGL3 to PLKO Cla1 For:5′⁃CTCGACGGTATCGATCACGAG⁃AC⁃3′,U6 array seq Rev⁃new:5′⁃CTGCCATTTGTCT⁃CGAGGTCGAG⁃3′,扩增产物大小为549 bp,利用pGL3 to PLKO Cla1 For引物进行测序。VDR基因编辑位点扩增引物为hVDR lib⁃testF:5′⁃GCTGAAG⁃CAGGAGGATTACTTGA⁃3′,hVDR lib⁃testR:5′⁃GA⁃CACCAACACACAGATCCTCAA⁃3′,扩增产物大小为717 bp,利用hVDR lib⁃testF引物进行测序。
1.3 统计学方法
实验数据利用GraphPad Prism 7.0统计分析,数据以均数±标准差()表示,采用单因素方差分析,使用Tukey 检验进行两组比较,P<0.05 为差异有统计学意义。
2 结果
2.1 可诱导表达Cas9载体的构建和验证
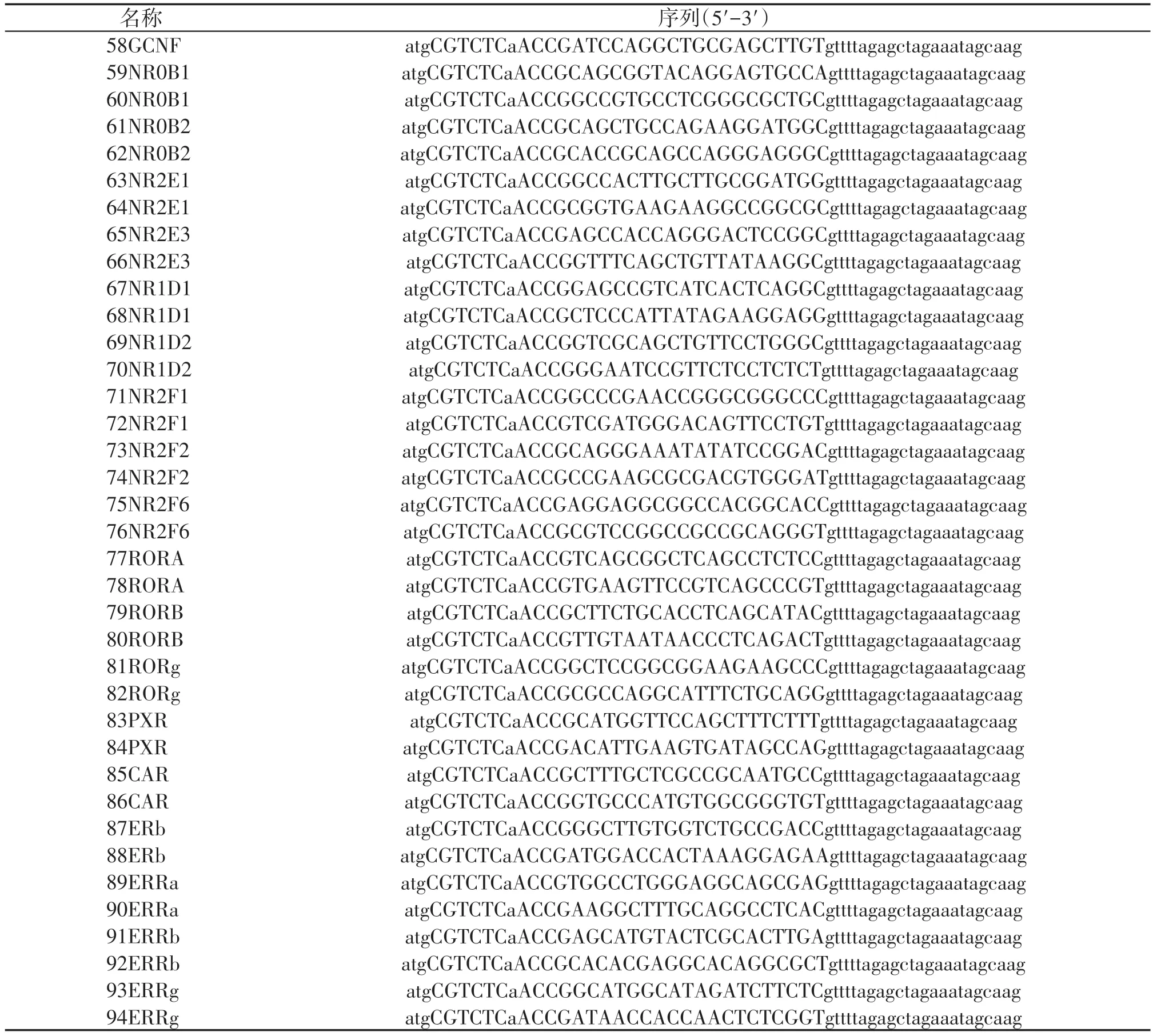
来自不同实验室的Cas9 载体各不相同,有的NLS放置在Cas9的一端[10-11],有的放置在两端[9],有的利用了不同的NLS 序列[9,11],密码子优化的规则也各不相同[9-12]。我们将不同版本的Cas9 和PGL3⁃U6⁃Rag1⁃sgRNA转染HEK293T细胞,利用T7EN1酶切来检测不同版本Cas9 的切割效率。结果显示,NLS⁃Flag⁃Cas9⁃NLS 和Cas9⁃NLS 具有相对较高的切割效率,光密度定量分析也得出一致的结果,但不同Cas9版本之间的切割效率差异无统计学意义(图1A、B)。我们选择NLS⁃Flag⁃Cas9⁃NLS 和Cas9⁃NLS这2 个版本的Cas9 进行PiggyBac 转座子载体的构建,通过连接IRES⁃hrGFP 来指示Cas9 的表达情况,通过连接BleoR 抗性基因,利用Zeocin 药筛来获得稳定插入Cas9 的细胞(图1C)。在瞬时共转染PB⁃TRE⁃NLS⁃Flag⁃Cas9⁃NLS 和PB⁃CAG⁃rtTA 的HEK293T 细胞中加入Dox,可以成功诱导hrGFP 的表达,而未加入Dox 的对照组中未见阳性信号(图1D)。为了进一步检测Cas9是否表达,我们利用an⁃ti⁃Flag抗体进行染色,结果显示GFP阳性的细胞,同时也是Flag 阳性的,并且Flag 信号定位在细胞核中(图1E),说明Cas9 蛋白成功表达和定位。有趣的是,Flag 染色结果显示Cas9 蛋白在核仁中富集,这和之前报道的现象一致[7],最近的研究表明,Cas9含有核仁定位信号,突变核仁定位信号会导致Cas9蛋白均匀分布在细胞核中,但是其切割效率也会随之降低[13]。
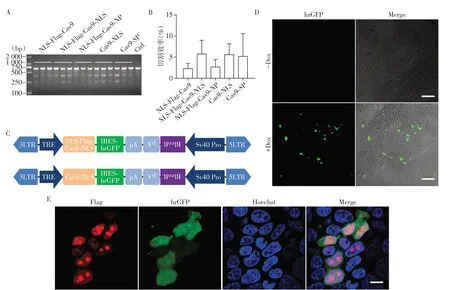
图1 可诱导表达Cas9载体的构建和验证Figure 1 Construction and verification of inducible Cas9 expression vector
2.2 可诱导表达Cas9的稳定细胞系的构建和验证
为了提高转座效率,我们使用插入片段较小的PB⁃TRE⁃Cas9⁃NLS⁃IRES⁃hrGFP⁃Zeo 载体构建可诱导表达Cas9 的稳定细胞系。PiggyBac 转座子载体、PB⁃CAG⁃rtTA 和转座酶载体共电转,加入Zeocin 传代3次之后,挑选圆形的单克隆接种到96孔细胞板中继续培养,加入Dox 之后,在荧光显微镜下挑选GFP 阳性的克隆,共挑出7 个GFP 信号较强的克隆(1A、1G、2B、3G、5H、8E、10A)继续培养(图2A)。为了挑选效率最高的细胞系,我们在这7 个细胞株中电转了PGL3⁃U6⁃Rag1⁃sgRNA,PCR扩增目的片段并进行T7EN1酶切,结果显示8E克隆具有较高的切割效果(图2A)。为了进一步检测切割效率,我们将PCR 产物进行了TA 克隆并测序,在17 个成功测序克隆中检测到15 个克隆含有突变,效率高达88.2%(15/17),其中76.5%(13/17)的克隆发生了碱基缺失,11.8%(2/17)的克隆同时发生了缺失和插入(图2B),这些结果说明我们成功构建了可诱导表达Cas9的稳定细胞系。
图2 可诱导表达Cas9的稳定细胞系的构建和验证Figure 2 Generation and verification of a stable HeLa cell line with inducible expression of Cas9
为了检测HeLa 8E细胞株在其他基因位点的基因编辑效果,我们针对人ERb、ERRa、ERRb和ERRg 4 个基因设计相应的sgRNA,并检测切割效率。传统的sgRNA 表达载体构建依赖oligo 的退火然后连接到载体中,无法进行sgRNA表达载体的高通量构建,为了能在HeLa 8E细胞上进行高通量筛选,我们将sgRNA识别序列设计在前向引物上,以sgRNA的骨架为模板,和公共后向引物扩增出携带完整sgRNA 序列的片段,利用二类限制性内切酶Esp3Ⅰ和T4 DNA连接酶,通过边切边连的方式,可以高通量地构建sgRNA 表达载体(图2C)。我们利用这种方法一次性构建了8 条sgRNA 表达载体,针对每个基因,等质量混合2 条sgRNA 载体,并电转HeLa 8E细胞系,PCR 扩增结果显示每个基因都出现了明显的片段缺失(图2D)。以上结果证明我们构建的可诱导表达Cas9 的稳定细胞系没有编辑位点的偏好性,可以在多个基因位点进行高效基因编辑,为下一步用于高通量sgRNA文库筛选打下了很好的基础。
2.3 顺铂耐药性核受体基因的筛选
顺铂是一种含铂的抗癌药物,对恶性上皮肿瘤、淋巴瘤等都有治疗功效,长期使用这种药物会导致人体产生耐药性,但是耐药机制仍存在争议[1]。核受体超家族是配体激活的转录因子,共含有48个基因,在细胞分化、增殖和代谢中发挥不同作用,参与多种癌症的发生、发展[14]。核受体是重要的药物靶点,阻断乳腺癌患者雌激素受体(ERa)作用的药物或阻断前列腺癌患者雄激素受体(AR)作用的药物显著改善了患者的生存,但也伴随着严重的药物耐药性[14]。为了探究核受体超家族成员是否参与顺铂耐药性的发生,我们针对目前鉴定的47个人类核受体基因(由于HeLa 细胞不表达ERa[15],所以排除此基因),设计了94 条sgRNA,组成了一个小型的sgRNA 文库(图3A),包装成病毒颗粒之后感染HeLa 8E 细胞,利用嘌呤霉素筛选整合进sgRNA 载体的细胞。在开始顺铂耐药性实验前48 h,加入Dox以诱导Cas9的表达和对目标基因的编辑。我们设计了3 个顺铂浓度梯度,在对照细胞全部死亡之后,撤掉顺铂。从0.9 μg/mL 和1.5 μg/mL 两组中共长出37 个单克隆(图3B),挑选每个单克隆并扩增培养,提取基因组DNA之后PCR扩增包含sgRNA的序列,Sanger 测序结果发现21 个克隆含有维生素D受体(vitamin D3 receptor,VDR)sgRNA,2 个克隆含有维甲酸受体β(retinoic acid receptor beta,RARb)sgRNA,含有视黄醇X 受体(retinoid x receptor beta,RXRb)和类固醇生成因子⁃1(nuclear receptor 5A1,NR5A1)sgRNA 各有1 个克隆(图3C),剩下12 个克隆sgRNA 位点显示乱峰,无法确定具体是哪个sgRNA。为了进一步验证目标基因是否被编辑,我们对21个含有VDR sgRNA单克隆的VDR区域进行了测序,结果显示20个克隆的sgRNA识别区域发生了突变(图3D)。

图3 顺铂耐药性核受体基因的筛选Figure 3 Screening of cisplatin⁃resistant nuclear receptor genes
3 讨论
本研究利用PiggyBac 转座子系统,成功构建了可诱导表达Cas9 的稳定HeLa 细胞系,并证明在多个位点可以高效切割基因组。同时,我们针对人核受体基因构建了sgRNA文库,在这个细胞系上进行了顺铂耐药性的筛选。之前有研究表明,PiggyBac转座子在转座酶存在的情况下会发生再转座或者丢失的现象[16],为了防止这种情况的出现,我们转座酶是瞬时表达的,不会持续性存在;同时,在培养HeLa 8E 细胞株的过程中,也会定期加上Zeocin 进行药筛,以排除Cas9丢失和被沉默的可能。除了通过诱导控制Cas9 的表达,Cas9 切口酶/双sgRNA[7]、截短sgRNA[17]、高保真Cas9 突变体[18-23]等策略也用于降低CRISPR/Cas9的脱靶效应。另外,多个比sp⁃Cas9小的同系物如saCas9[24]、Cpf1[25]等也被发现,一定程度上解决了转导效率低的问题。关于Cas9 在核仁中富集,之前的研究表明,一旦表达sgRNA,Cas9 会均匀分布在细胞核中[7],这种现象是很难用Cas9 含有核仁定位信号来解释的[13],可能是由于Cas9/sgRNA复合体的溶解性更好,亦有可能是其他机制导致,值得进一步的研究。
之前有文章报道核受体基因ERb 的激动剂可以调节胶质母细胞瘤细胞对顺铂的敏感性[14],鼓励我们去探究核受体超家族成员与顺铂耐药性的关系。在筛选的过程中,由于含有RARb、RXRb 和NR5A1 sgRNA的克隆较少,我们没有进一步去验证对应的基因是否被编辑,以及有12个克隆含有多个sgRNA,也没有进一步去验证具体是哪些基因被编辑,这些都值得继续探索。20 个含有VDR sgRNA克隆的测序结果显示,所有单克隆都携带相同的突变,提示这20 个单克隆可能来源于同一个细胞,之前有文章报道维生素D3及其受体VDR调控机体的钙磷代谢稳态,对多种肿瘤细胞的增殖有抑制作用。因此,突变VDR 可能会使HeLa 8E细胞的增殖加快,导致长出的克隆大部分含有VDR突变,当然,这也可能是顺铂耐药性发生的原因,需要进一步通过直接敲除或者过表达VDR进行正反验证,具体的机制也值得我们深入去解析。
