肝豆状核变性的基因诊断研究进展
2021-11-05姚碧莲张欣欣
姚碧莲, 张欣欣, 韩 悦
(上海交通大学医学院附属瑞金医院a.全科医学科;b.临床病毒研究室,上海200025)
肝豆状核变性又称威尔逊病(Wilson disease,WD),是由于ATP酶铜转运β基因ATP7B[1-2]发生变异、造成体内铜代谢紊乱的常染色体隐性遗传病。该基因的致病变异导致ATP酶的功能缺陷或丧失,造成胆道排铜障碍,大量铜蓄积于肝、脑、肾、骨关节、角膜等组织和脏器[3-5]。本病在世界范围内携带者频率为1∶100~1∶90,流行率在1∶40 000~1∶30 000之间[6]。既往国外报道WD人群发病率为1/10万~1/3万,杂合子频率为1/200~1/100[7],而英国一项研究认为其理论发病率不低于1/7 026,推测外显率低下和诊断的局限性可能是实际发病率统计偏低的主要原因[8]。胡文斌等在安徽省3个县连续进行2次调查,共调查153 370人,发现WD患者9例,推测患病率为5.87/10万[9-10]。一 般 认 为 该病发病率在中国比西方国家更高[11]。
由于铜沉积的组织器官不同,因此临床上WD患者临床表现多种多样,临床表现既往可主要分为肝型、脑型、混合型和其他类型。肝型患者主要临床症状包括无症状性转氨酶升高、肝肿大、脾肿大、肝炎、脂肪肝、肝硬化和急性肝衰竭等。脑型患者表现主要包括帕金森综合征,运动障碍,口、下颌肌张力障碍,精神症状等。因过量的铜还会沉积于肾脏、骨骼关节、血液、皮肤、角膜等其他组织器官中,引起相应的组织器官损伤,可表现为蛋白尿、血尿、全身水肿、骨折、关节疼痛、心肌损害、肌病、溶血性贫血、皮肤色素沉着、角膜色素环(Kayser-Fleischer ring,K-F环)等[12]。临床分型更为全面的有潜伏型(症状前期型)、脑型、内脏型、脑-内脏型、骨-肌型、脊髓型/脑脊髓型[13]。中国最新发布的《中国肝豆状核变性诊治指南2021》着重将复杂临床表现按肝损害、神经精神损害、其他系统损害及症状前个体进行详细叙述[14]。
WD目前诊断依靠临床症状[神经和(或)精神症状、原因不明的肝损害、K-F环阳性]、溶血、生化检查[血清铜蓝蛋白降低和(或)24 h尿铜升高]以及ATP7B基因检测[15]。WD患者临床表现复杂多样,可累及各个系统,早期临床症状多样且不典型,极易被误诊或漏诊,生化检查存在假阳性、假阴性,早期诊断较为困难。肝脏组织铜定量检测虽然有较高的特异性和诊断价值,但该检查为有创操作,存在风险。因此基因检测对于患者的筛查、早期诊断和精确诊断有重要意义。
ATP7B基因及突变
研究认为,WD是双等位基因变异引起的单基因缺陷疾病。ATP7B基因定位于13q14.3,有21个外显子,编码区长4.1 kb。已知突变主要集中在几个热点区域,同时存在大量罕见突变。到目前为止,人类基因突变数据库(HGMD,www.hgmd.org)报道了907个不同的有害变异,通过编码外显子和内含子序列分析,可检测到878个变异(96.8%)[16],还有很多新的突变不断被认识。其突变类型很复杂,包括错义、无义、缺失、插入、剪切位点突变,其中由单核苷酸引起的错义或无义突变最常见(60%),其次是插入、缺失(26%)和剪切位点突变(9%)[17]。p.H1069Q突变是西方人群较常见的突变之一,大多以复合杂合形式出现,合并的突变不同[18]。我国WD患者主要有p.R778L、p.P992L和p.T935M 3个高频致病突变,占所有致病突变的50%~60%[19-22],其他7种常见突变包括p.A874V、p.I1148T、p.Q511X、p.N1270S、p.G943D、p.R919G和p.R778Q,可占所有致病突变的67%[20]。由于不断有新的突变被发现,ATP7B基因突变数据库是动态更新的。杨任民等[23]在1997年率先报道了国内WD患者新的变异位点S662T;我院张欣欣课题组研究总结了我国WD的基因型-临床表型特点,并发现扩充了14个新的突变[24]。另外有长期研究在2004—2015年中国632例WD患者中发现173个ATP7B变异,其中58个是新发现的变异[25]。
基因诊断策略
一、疑似患者基因检测
针对临床疑似患者,基因检测方法包括对21个外显子编码区域及其相应邻近的内含子区域序列的分析,可以根据当地人群中突变频率的不同,将突变热点区域的外显子优先排序。如果使用一代测序方法不能发现ATP7B外显子编码区域和邻近内含子序列存在已知致病相关变异或突变,临床仍高度怀疑,则可考虑启用二代测序(next generation sequencing,NGS)技术,包括全外显子测序(whole-exome sequencing,WES)或全基因组测序(whole-genome sequencing,WGS),对ATP7B基因进行更全面的测序(可包含启动子和内含子区域的序列),或寻找包括ATP7B基因在内的其他肝病相关的基因是否存在罕见、未知突变或其他结构改变,并结合家系基因型-临床表型或实验数据判断致病性,扩充WD致病相关基因突变库。NGS具有高通量检测特点,比一代测序能更全面地检测出基因突变,提高诊断效率。在北美和欧洲儿科胃肠病学、肝病学和营养学会的最新指南已经推荐在合适的情况下使用NGS检测基因突变[26]。随着成本的控制,NGS技术在临床工作中有更多的应用机会。
二、WD患者家系筛查
针对确诊WD患者进行家系筛查至关重要。患者兄弟姐妹是纯合子的概率是25%,患者子女是纯合子的概率是0.5%。对于家属的基因检测,可以通过一代测序分析患者所示的致病突变基因或者单倍型分析,如发现致病突变基因或致病单倍型,可协助诊断WD(见图1),及时予以治疗。
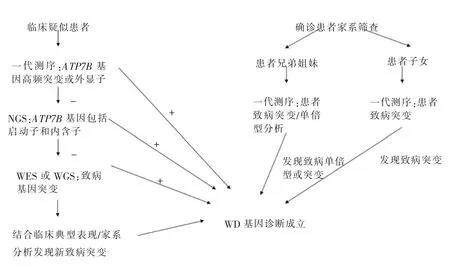
图1 WD基因检测对象及策略
我国2021年新版指南提醒临床医师应高度警惕血清铜蓝蛋白<120 mg/L的个体及肝酶升高且24 h尿铜≥40 μg儿童,建议进行ATP7B基因检测以明确诊断[19]。同时更新了WD的基因筛查策略,指出临床医师应先针对性筛查我国WD患者的ATP7B基因高频致病突变,有助于节约医疗资源。对于未检出高频致病变异的患者,进一步筛查ATP7B基因全长编码区及其侧翼序列的致病变异以助于确诊。
ATP7B基因突变与基因型-表型的关系分析
目前,众多研究集中在探索基因型和表型之间的关系,并尝试解释其复杂多样的临床表现。基于人群样本的一项研究表明,患者在血铜蓝蛋白水平较低的情况下,p.R778L纯合子突变患者较杂合子突变患者肝损害表现明显,即p.R778L突变对ATP7B功能影响较大[27-28]。有细胞功能实验证明,p.R778L突变与p.T935M和p.R919G突变相比对功能影响更大[29-30]。有研究认为p.R778L突变WD患者的发病年龄较晚,与首发症状和铜代谢障碍无关[31]。但是另一项针对中国人群的研究发现,p.R778L预示发病年龄更早,铜蓝蛋白及血清铜的水平更低,而p.T935M和p.R919G则与铜蓝蛋白水平较高有关。携带p.R919G的患者多以单纯神经系统症状为主,而携带p.T935M的患者则往往同时具有神经系统和肝脏病变的表型[21]。在有限的人群样本中,研究发现p.L492S、p.V519M、p.E541K、p.L549P、p.A604P、p.R616W、p.P992L、p.N1270S等往往预示着发病年龄较早,而具有p.I1148T突变的患者则大多在12岁后发病[32]。另有研究表明,p.M769Hfs*26(c.2299insC)携带者肝脏疾病症状表现较多,带有p.H1069Q/p.M769Hfs*26复合杂合突变的患者常常具有相似的临床表型和发病年龄,表现为构音障碍及吞咽困难等症状[33]。而p.A1003T突变携带者与神经系统疾病之间关联更为密切[18]。还有文献报道,临床上有极少部分的WD患者并没有发现铜蓝蛋白水平降低,可能是位于第一个跨膜结构的p.S653Y突变阻止了ATP7B从反面高尔基网(trans-Golgi network,TGN)上的解离,但在此之前,其能够正常转运铜离子以供铜蓝蛋白的合成,因此造成了该少见的临床表型[34]。已报道的外显子长片段缺失突变有5种(Exon2-4deletion、Exon5-6deletion、Exon6-8deletion、Exon20deletion、Exon20-21deletion)[35]。在以往的研究中,携带大片段缺失的幼儿多存在较为严重的肝脏病变,如进行性肝炎、肝硬化、肝衰竭,但无神经系统症状[36](见表1)。

表1 ATP7B突变与临床表型
ATP7B基因单核苷酸多态性携带者发病分析
临床上有极少部分的患者仅检测出ATP7B基因中的基因单核苷酸多态性(single nucleotide polymorphism,SNP)位点。例如p.S406A、p.V456L p.K832R、p.R952K等,相对于野生型ATP7B的铜离子转运速率较低,但并未从根本上影响ATP7B基因功能[37]。在基因分析过程中,会常规过滤掉人群中常见的SNP位点,因而会漏掉许多功能性SNP。对于WD患者,需要在基因检测中注意上述这些位点的变异。
修饰基因对WD临床表型可能存在的影响
不同WD患者之间的临床表型差异较大,甚至在相同生活背景下的家系成员间也时常有所不同,因此不能排除其他基因参与了致病过程。铜代谢结构域包含性蛋白1(copper metabolism domain containing 1,COMMD1)是一种与铜代谢紊乱相关的蛋白,其第164位天冬氨酸的同义突变p.D164D是p.H1069Q纯合突变患者神经和肝脏症状早发的重要因素[38]。此外,存在超氧化物歧化酶2(superoxide dismutase,SOD2)基因p.V16A纯和突变的男性患者常常发病较早,而存在过氧化氢酶(catalase,CAT)基因c.262C>T纯和突变的男性患者发病较晚[40]。在印度人群中,有研究发现伴有神经症状WD患者的脑源性神经营养因子(brain derived neurotrophic factor,BDNF)大多存在p.Y90Y同义突变,且患者的平均发病年龄明显推迟,提示BDNF可能作为一种潜在的ATP7B相关修饰基因影响WD患者的表型[41]。还有研究者提出载脂蛋白E(apolipoprotein E,ApoE)和朊粒蛋白(prion protein,PRNP)的突变是影响WD患者临床表型的潜在因素,其突变可能与WD患者神经系统表型相关[42-45]。Kluska等[45]对248例WD患者进行了全外显子组测序分析,该研究确定了酯酶D(eaterase D,ESD)和染色体重塑因子INO80(INO80 complex subunit)2种新的可能变异,分别与神经症状风险的增加和降低有关。
其他已报道的WD修饰基因还包括人铜转运蛋白-1(human antioxidant 1 copper chaperone,ATOX1)[46]、X连锁凋亡抑制蛋白(X-linked inhibitor of apoptosis,XIAP)[47]、patatin样磷脂酶域蛋白3(patatin like phospholipase domain containing 3,PNPLA3) 基 因[48]、 亚 甲 基 四 氢 叶 酸 还 原 酶(5,10-methylenetetrahydrofolate reductase,MTHFR)[49](见表2)。

表2 修饰基因突变与临床表型
另外,在表观遗传学领域,有文献报道[50],全基因组重亚硫酸盐测序在人类肝脏样本中发现了969个高甲基化和871个低甲基化差异性甲基化区域(differentially methylated region,DMR),其中包括18个具有全基因组意义的区域。WD患者肝脏的DMR与叶酸、脂质代谢和急性炎症反应等功能丰富的基因相关,并能区分WD患者早期和晚期纤维化。通过检测血液中DMR可区分肝型WD患者组、健康组和疾病对照组,并可区分肝型WD和脑型WD。这提示了表观变化对致病性的影响,给未来的临床诊断带来更多的思路和方法。
总结和展望
由于WD可以在任何年龄起病,症状复杂多变且隐匿,给临床早期诊断带来了很大的困难。基因检测对于患者的筛查、早期和精确诊断有重要意义。中外基因突变谱系、热点突变优先排序往往存在差异,而且ATP7B基因突变数据库是动态更新的,需要持续与国际、国内数据库进行对接。目前的研究难点在于除少数热点突变外,多数突变在人群中的频率极低,一般难以收集到足够的病例进行统计分析,而ATP7B基因型-表型依赖大量临床数据积累,进而在功能学实验基础上可明确相应致病性并辅助诊断。另外,除了ATP7B基因本身,其他遗传因素也可影响临床表现,其中表观遗传因素对WD表型异质性的作用有待进一步研究证实。
