牛亚科物种TRs分布特点及着丝粒区卫星DNA进化研究
2021-11-04张天留范婷婷张路培徐凌洋高会江李俊雅
张 瑞,马 钧,陈 燕,张天留,范婷婷,朱 波,张路培,徐凌洋,高会江,李俊雅,高 雪
(中国农业科学院北京畜牧兽医研究所,牛遗传育种创新团队,北京 100193)
串联重复序列(tandem repeats sequence,TRs)是指核心重复单元以首尾相连的方式多次重复所组成的序列,对细胞有丝分裂和减数分裂过程中染色体的分离及结构稳定起到至关重要的作用[1-2]。根据重复单元的长度不同,TRs可分为3大类,即卫星DNA(satellite DNA)(>100 bp)、微卫星DNA(microsatellite DNA)(≤6 bp)和小卫星DNA(minisatellite DNA)(7~100 bp)[3-4]。微卫星和小卫星DNA为中度串联重复,主要位于基因组非编码区、内含子和常染色质区域,具有高度多态性,因此常被用来构建个体DNA指纹图谱[5]、评估遗传多样性[6-9]、分析种群结构[10-11]或鉴定个体间亲缘关系[12-14];而卫星DNA为高度串联重复序列,是异染色质的重要组成部分,主要集中在着丝粒区、端粒区附近以及Y染色体上[15-16]。研究表明,着丝粒区的卫星DNA参与牛科物种染色体融合[17];卫星DNA序列变化加速物种和群体分化[18];因此,可用卫星序列在组成、物理位置的变化来推断物种及染色体的进化关系[19-20]。
TRs的碱基改变、扩张收缩和大片段复制都会影响着丝粒和近着丝粒区序列快速进化[21]。Melters等[22]利用生物信息学方法对不同物种的TRs进行比较分析,结果表明,几乎所有动植物基因组的着丝粒区都存在高拷贝的卫星序列,且在物种间快速进化,但当分化时间超过5 000万年,其序列相似度迅速降低。1978年,Macaya等[23]利用密度梯度离心法从牛基因组中分离出包括1.709、1.715、1.723、1.720a、1720b、1.711a、1.711b、1.706在内的8种卫星DNA,其中1.720、1.711、1.706卫星序列相似性较高。1982年,Taparowsky和Gerbi[24]提出了卫星序列进化的假设模型,并将卫星DNA分成了A、B两大家族,家族A包括1.706、1.711a、1.720,家族B则包括1.715、1.711b。1996年,Modi等[25]对偶蹄目下46个物种分析发现,着丝粒区特异性卫星序列-1.715卫星家族和bovine-Pst广泛存在反刍动物中。Kopecna等[26]通过激光显微技术研究了10个牛科种群着丝粒区卫星DNA-1.715,并分析了它们之间的亲缘关系,发现野牛和普通牛、非洲水牛和亚洲水牛4个物种亲缘关系更加紧密。2013年,Melters等[22]对282个动植物基因组研究发现,着丝粒区存在大量的TRs;瘤牛、普通牛、水牛、欧洲野牛等物种着丝粒区存在1 410 bp(1.715)和680 bp(1.723)卫星家族,但两者序列无相似性,且前者的密度更小,丰度更低;普通牛中,1 410 bp 序列达到了71%,而680 bp序列仅为29%。
目前,已报道的牛亚科基因组有普通牛、独龙牛、非洲水牛、欧洲野牛等,它们的重复序列占比分别为48.81%[27]、48.13%[28]、37.21%[29]、47.03%[30]。2009年,Adelson等[31]分析了普通牛基因组中转座子和简单重复序列(simple sequence repeats,SSR)的分布和特征,发现其与GC含量和基因密度等有一定的相关性。2012年,赵芳芳[32]研究了牦牛部分基因组(占全基因组9.51%)中微卫星重复序列的特征。但目前关于串联重复序列特征研究较少,且在普通牛及其他牛亚科物种中尚未有相关报道。本研究基于普通牛、瘤牛、水牛、牦牛、野牛、独龙牛6个 牛亚科物种的基因组序列,通过TRF和RepeatMsker 软件鉴别了6个牛亚科物种基因组中的TRs,研究了TRs在其基因组中的组成、分布及结构特征,并分析了6个牛亚科物种染色体着丝粒区卫星序列的进化,为牛亚科物种TRs的研究提供理论和数据支撑,也为进一步研究牛亚科物种的进化提供重要依据。
1 材料与方法
1.1 数据收集
本研究以普通牛、瘤牛、牦牛、水牛、野牛和独龙牛的基因组序列为研究对象,其中普通牛、瘤牛、牦牛、水牛和野牛的参考基因组序列来源于NCBI基因组数据库(https://www.ncbi.nlm.nih.gov/genome/),下载版本分别为普通牛(Bostaurus, ARS-UCD1.2)、瘤牛(Bosindicus, Bos_indicus_1.0)、水牛(Bubalusbubalis, UOA_WB_1)、野牛(Bisonbison, Bison_UMD1.0)、牦牛(Bosmutus, BosGru_v2.0);独龙牛基因组序列使用本团队组装版本。
1.2 串联重复序列的鉴定
通过TRF(tandem repeats finder,V4.09)[33]和RepeatMasker(V4-0-9)两个软件对串联重复序列进行鉴定,重复单体的长度在1~2 000 bp之间。1)TRF:参考Melters等[22]的方法,具体参数设置为1、1、2、80、10、200、2 000,分别表示匹配(match)、插入缺失(indel)、匹配概率(probability of match,PM)、插入缺失的概率(probability of indel,PI)、最大周期(maxperiod)、错配(mismatch)、最低得分(minscore)。2)RepeatMasker:将不同物种的基因组分别与数据库(Dfam_Consensus和RepBase)中的序列进行比对查找。运行命令为time RepeatMasker -parallel 2 -species ‘name’ -gff -dir repeat fasta_sequence,输入文件格式为fasta序列,其中-parallel 2表示并行的线程数为2,-species ‘name’表示所对应物种的名字为’name’,-gff 表示输出格式为gff,-dir repeat表示将结果输出repeat文件夹中。3)数据整合与处理:将TRF和RepeatMasker两个软件鉴定的TRs进行整合。本研究中对于重叠部分的序列,只保留重复单元较短的序列。
1.3 串联重复序列的分类
根据重复单体的长度,将TRs分为3类:1)卫星DNA:重复单体长度>100 bp;2)小卫星DNA:重复单体长度在7~100 bp之间;3)微卫星DNA:重复单体长度≤6 bp。
参考Castoe等[34]的方法,具体为:对于单碱基、二碱基、三碱基、四碱基微卫星,总长度≥12 bp;对于五碱基和六碱基微卫星,其总长度≥15 bp。根据起始碱基顺序差异和碱基互补配对原则,对不同的拷贝类别进行归类处理。例如单碱基A及其互补碱基T归为同一类;二碱基重复单元AC、CA其互补序列TG、GT归为同一类;三碱基重复单元AAC、ACA、CAA及其互补序列TTG、TGT、GTT归为同一类。
1.4 着丝粒区卫星序列的提取
根据Melters等[22]的报道,牛亚科物种着丝粒区卫星DNA主要有1.723(680 bp)和1.715(1 410 bp)两类。因此,本研究主要对1.723和1.715卫星DNA进行分析。1)卫星DNA下载:从NCBI中的核酸数据库(https://www.ncbi.nlm.nih.gov/nucleotide/)中下载这两个序列,登录号分别为M36668.1和J00036.1;2)建库:建立6个物种基因组序列的本地数据库(makeblastdb -in fasta -parse_seqids -hash_index -dbtype nucl);3)比对:将1.723和1.715卫星序列分别作为参考序列,结合本地BLAST进行核酸比对(Blastn);4)候选序列提取:根据比对结果中卫星序列的位置信息,利用 Perl程序提取得分> 800的序列作为候选卫星序列,进行后续分析。
1.5 系统发育树的构建
首先,将6个物种基因组中的1.723和1.715卫星序列整合到一个文件中;然后,利用ClustalW程序进行序列比对,采用默认参数,输出结果为phylip格式;之后,利用 Phylip软件构建系统发育树(NJ树),重复1 000次,其他为默认参数,输出文件为tree文件;最后,利用FigTree软件(V1.4.3,http://tree.bio.ed.ac.uk/software/figtree/)可视化进化树。
2 结 果
2.1 牛亚科物种TRs比较分析
通过TRF和RepeatMasker软件鉴定出单元长度在1~2 000 bp的TRs,并将其分为微卫星DNA、小卫星DNA和卫星DNA进行比较分析(表1)。由表1可知,TRs在6个物种基因组中的平均占比为2.03%,平均长度为54.93 Mb,其中普通牛所占比例最高,3.42%(93.00 Mb),瘤牛比例最低1.42%(37.88 Mb)。这些TRs中,微卫星DNA在6个物种基因组中的平均比例略高,为0.76%(20.46 Mb),而小卫星DNA和卫星DNA在基因组中的比例相差不大,分别为0.65%(17.69 Mb)和0.62%(16.78 Mb)。
同时,本研究还统计分析了每个物种基因组中TRs位点数,如表2所示,6个物种基因组中,TRs总位点数为523 165~592 305,均值为564 611。微卫星DNA在基因组中分布最广泛,位点数最多(453 378~508 830),占其总数的85.64%。小卫星和卫星DNA在基因组中的位点数较少,其均值分别为43 026(7.62%)和38 180(6.75%),但每个位点的平均长度却明显高于微卫星DNA(图1),表明重复单元较短的序列每个位点的重复序列长度可能也较短。

图1 串联重复序列平均每个位点的序列长度Fig.1 The sequence length of each locus in tandem repeats sequence
2.2 卫星DNA的分布特点
由表1、表2可知,卫星DNA在6个牛亚科物种基因组中平均长度为16.78 Mb,其中独龙牛的卫星DNA最长,为24.65 Mb,野牛则最短,为11.86 Mb;而卫星DNA在6个牛亚科物种基因组中平均位点数为38 180(6.75%),其中独龙牛卫星DNA位点数最多,为46 908(8.09%);野牛的位点数最少,为31 144(5.53%),略低于6个物种卫星DNA位点平均比率6.75%。这表明,在卫星DNA中,基因组中的位点数越多,其序列长度也越高。
2.3 小卫星DNA的分布特点
由表1、表2可知,小卫星DNA在6个牛亚科物种中比例为0.26%~1.98%,平均位点数为43 026,占TRs位点总数7.62%。6个物种中小卫星DNA位点数相差不大,其中位点数最多的是野牛,为45 349, 占8.06%;而瘤牛最少,为37 708,占比7.21%。6个物种小卫星DNA平均长度为17.69 Mb,占0.65%,其中瘤牛小卫星DNA长度最短,为7.07 Mb,占瘤牛基因组总长的0.26%;普通牛则最长,为53.79 Mb,占普通牛基因组总长的1.98%。表明小卫星DNA长度在6个物种间变化较大,可能是由物种间差异造成的,也有可能与不同基因组的组装效果有关。

表1 串联重复序列在6个牛亚科物种基因组中的长度及比例

表2 串联重复序列在6个牛亚科物种基因组中的位点数及比例
2.4 微卫星DNA分布特点
由表1、表2可知,微卫星DNA在6个牛亚科物种中的比例为0.67%~0.85%,总长度在18.03~23.05 Mb之间,总数量在50万左右,远远大于小卫星和卫星DNA;其中水牛的微卫星位点数最高(508 830个),其次是普通牛(490 638个),牦牛的数量最少,为474 360个。通过对一至六碱基微卫星序列的丰度和长度分析发现,6个牛亚科物种中,二碱基微卫星序列的丰度最高(图2a),在基因组中分布最广泛;三、五、六碱基微卫星则相应较低,这与其他真核生物中的结果一致[35]。其中,二碱基微卫星DNA丰度在牦牛中最高,为70.93 loci/Mb,但平均位点长度以普通牛的六碱基最高(图2a, 2b)。
在单碱基微卫星中,A/T的含量高于G/C的含量。在二碱基微卫星中,牛亚科基因组中丰度最高的

loci/Mb表示每Mb序列所对应的重复序列位点数,bp/locus表示每个位点的序列平均长度;图中红色越深表示相应的值越高,蓝色越深表示相应的值越低;1~6分别代表单碱基、二碱基、三碱基、四碱基、五碱基和六碱基。下同loci/Mb represents the locus number of microsatellites per Mb, and bp/locus represents the average sequence length of each locus. In the figure, the deeper the red, the higher the value, the darker the blue, the lower the value. 1-6 represents mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, hexanucleotide, respectively. The same as below图2 微卫星丰度(a)和每个位点的平均长度(b)Fig.2 The abundance of microsatellites(a) and the average length of each loci(b)
是AC拷贝(31.51~38.16 loci/Mb)(图3a),然后依次是AT、AG、CG,这与人及其它哺乳动物研究结果一致,在植物中以AT最丰富[36]。同时,由图3b看出,AT拷贝的位点平均长度(52.79~56.53 bp/locus)均高于其他3种,尤以普通牛最高;而且无论是丰度还是位点平均长度,AT拷贝均高于CG。这可能与AT由两个氢键相连,而CG由3个氢键连导致微卫星的复杂度增加相关。

图3 二碱基微卫星丰度(a)和每个位点的平均长度(b)Fig.3 The abundance of dinucleotide microsatellites(a) and the average length of each loci(b)
图4显示,在三碱基微卫星中,TAA为重复最多的拷贝类别(4.91~6.17 loci/Mb),该类别在其他脊椎动物中出现的频率也较高[35]。而每个位点的平均长度,普通牛的TAG(82.73 bp/locus)和GCC(73.62 bp/locus)高于其他拷贝类别。四碱基微卫星中,AAAT和AAAC的丰度最高,而CGAT和ACCT的平均长度高于其他类型。五碱基和六碱基微卫星中,含量较高的分别为TTTAT和TATACA。在不同的重复类型中,丰度高的拷贝类别其平均长度不一定处于较高水平,该结果与Adams等[35]对71个脊椎动物的研究结果类似。在一至六碱基微卫星中,GC的比例低于AT,这可能由微卫星的丰度和密度与GC含量呈负相关而引起的[37]。
2.5 牛亚科物种着丝粒区卫星DNA的进化分析
在牛亚科物种的卫星序列中,着丝粒区卫星DNA在基因组中高度保守且具有物种特异性,主要包括1.723和1.715序列,后者广泛存在于反刍动物中[25]。因此,本研究分别构建了卫星序列1.723和1.715在6个物种中的系统发育树,如图5a和5b所示。由图5a可见,1.715卫星DNA普遍存在于6个牛亚科物种中,序列在物种间存在分化,但分化不明显。普通牛、独龙牛、水牛各自的1.715卫星序列较明显聚在一起,表现出物种的特异性。由图5b可见,卫星序列1.723在牦牛中不存在,而其他5个 物种明显分成两支,独龙牛和野牛聚在一起,普通牛、瘤牛、水牛聚在一起,但每个物种表现出较明显的物种特异性。表明即便在近缘物种中,着丝粒卫星DNA也是不断进化的。
为进一步了解着丝粒区卫星DNA在同一物种中不同染色体上的进化情况,本研究分别构建了普通牛和瘤牛不同染色体上1.715卫星DNA的系统发育树,如图5c和5 d所示。由图5c和5d可以看出,普通牛1.715序列主要分布于2、4、11、15、19号染色体上,而瘤牛则主要分布在2和7号染色体上,而且发现在普通牛的2和4号染色体,瘤牛的2和7号染色体仍存在一些共享序列。由此表明,即便是同一物种,卫星序列在不同染色体上既发生着进化,也存在着共享。
3 讨 论
串联重复序列在过去被认为是“垃圾”、“自私”、“寄生”的DNA,但随着越来越多的研究表明,TRs对物种进化、基因遗传变异、转录调控等具有重要的意义[38],在基因组中扮演着重要的角色[39]。本研究分析了6个牛亚科基因组中TRs的分布特点,并着重研究了微卫星1~6碱基重复单元的序列特征以及着丝粒区卫星序列的进化。结果表明,TRs在6个物种基因组中的平均占比为2.03%,总位点数为564 611,平均长度为54.93 Mb,其中以微卫星含量最高,分布最广,微卫星(483 405/85.64%)>小卫星(43 026/7.62%)>卫星序列(38 180/6.75%)。其中水牛的微卫星数量最高,为508 830个,其次是普通牛(490 638个),牦牛的微卫星数量最少(474 360个)。 赵芳芳[32]研究了牦牛部分基因组(占全基因组9.51%)中微卫星重复序列的特征,发现在9.51%牦牛基因组中有43 409个微卫星位点,全基因组则约有微卫星位点456 456个,本研究结果与其基本一致。Wang和Glanzmann等[28-29]通过TRF鉴定出独龙牛的串联重复序列含量为0.62%、非洲水牛为1.41%,而本研究中独龙牛串联重复序列含量2.1%,非洲水牛为1.96%,整体占比偏高。可能与基因组组装情况及重复序列统计方法有关,由于TRs的序列相似性较高,给基因组测序和组装带来巨大困难,导致TRs在基因组中组装不完全,同时鉴定方法的不同,也会造成结果的差异。本研究使用自己组装的独龙牛基因组序列,组装指标Contig N50和Scaffold N50都较Wang等[28]发表的独龙牛基因组有较大的提升,这为鉴定出更多的TRs提供了可能。同时本研究使用TRF和RepeatMasker两个软件来鉴定和筛选TRs,会比单纯使用TRF软件鉴定出较多的序列。
早期研究表明,几乎所有动植物基因组着丝粒区都存在高拷贝的卫星序列,对细胞有丝分裂和减数分裂中染色体的分离及结构稳定性起到至关重要的作用[1-2]。Melters等[22]发现,在普通牛、瘤牛、欧洲野牛和水牛等物种着丝粒区普遍存在1.723(680 bp)和1.715(1 410 bp)两类卫星家族,且后者的丰度更高,密度更大;在普通牛中,1 410 bp序列占比达到了71%,而680 bp仅为29%。因此,本研究构建了1.723和1.715卫星DNA在6个牛亚科物种中的系统发育树,发现牛亚科6个物种基因组中均存在1.715卫星DNA,而牦牛中不存在1.723卫星DNA。1.715和1.723卫星DNA分布在不同的分支上,存在较明显的分化,表明即便在近缘物种中,着丝粒卫星序列也是在不断进化的,具有物种特异性,这与Fry等[40]的研究结果一致。普通牛、独龙牛、水牛各自的1.715序列较明显聚在一起,而其他几个物种的1.715序列变异较大。卫星序列1.723在普通牛、瘤牛、水牛、独龙牛和野牛5个 物种明显分成两支,独龙牛和野牛聚在一起,普通牛、瘤牛、水牛聚在一起,表明独龙牛和野牛的关系较近;而普通牛、瘤牛、水牛关系较近。这与Naji等[41]利用古等位基因AA和变异等位基因DA构建的9个牛科物种系统进化树结果较一致,独龙牛和班腾牛、印度野牛、美洲野牛关系较近。另外,卫星DNA一般不发生转座,但仍有部分卫星家族可以从一条染色体上传播到另一条染色体,使非同源染色体的着丝粒区卫星DNA高度相似[42]。研究发现,人类基因组中的卫星DNA可以在1、5和19号染色体,13和21号染色体以及14和22号等不同染色体上共享[43-45]。本研究构建了1.715卫星DNA在普通牛和瘤牛不同染色体上的系统发育树,发现1.715序列主要分布于普通牛的2、4、11、15、19号 染色体上,而瘤牛中则主要分布在2和7号染色体上,且2、7号染色体存在共享片段,这与人类的研究结果相似。
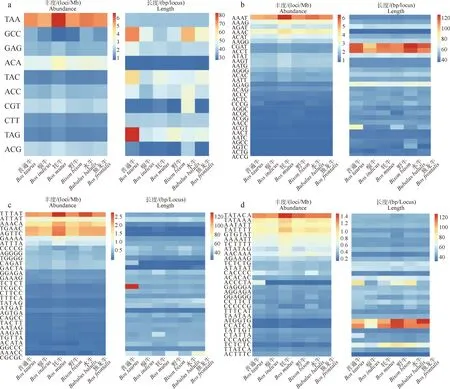
a、b、c、d分别表示三碱基、四碱基、五碱基和六碱基微卫星重复类型的分布,其中b、c、d选取排名前30的微卫星序列进行分析a,b,c,d represent distribution of trinucleotide, tetranucleotide,pentanucleotide, hexanucleotide, respectively, and the top 30 microsatellite sequences are selected for analysis in b,c,d图4 三至六碱基微卫星丰度和每个位点的平均长度Fig.4 The abundance of 3-6 base microsatellites and the average length of each loci
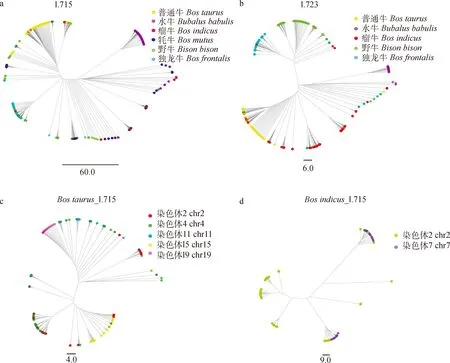
a和b分别表示1.715和1.723卫星DNA在不同物种间的进化,不同颜色代表不同物种;c和d分别表示1.715卫星序列在普通牛和瘤牛中的进化,不同颜色代表不同染色体a and b represent phylogenetic trees of 1.715 and 1.723 satellite sequence in different species,respectively, the different colors represent different species; c and d represent phylogenetic trees of the 1.715 satellite sequence in Bos taurus and Bos indicus, respectively, the different colors represent different chromosomes图5 牛亚科物种着丝粒区卫星DNA系统发育树Fig.5 Phylogenetic trees of centromeric satellite sequence in the 6 bovinae genomes
4 结 论
本研究发现,TRs在牛亚科6个物种中平均占比为2.03%,微卫星为TRs主导序列,占比85.64%;且二碱基微卫星丰度最高,并以AC拷贝类别为主;1.715卫星DNA普遍存在于6个牛亚科物种的基因组中,但在物种间或染色体间存在不同程度分化。本研究结果将为研究牛亚科物种间TRs的进化关系提供重要理论支撑。
