朝药桑黄的质量标准研究
2021-10-29王乐奇杨树东宋爱群
王 辉 王乐奇 杨树东 韩 达 宋爱群
1.长春市食品药品检验中心中药室,吉林长春 130012;2.东北师范大学化学学院,吉林长春 130024;3.长春市食品药品检验中心分析仪器研究室,吉林长春 130012;4.吉林省通化博祥药业股份有限公司项目部,吉林集安 134200
[关键字]朝药桑黄;微波消解-电感耦合等离子体质谱;重金属及有害元素;薄层色谱鉴别;质量标准
桑黄始载于《神农本草经》,《本草经集注》《药性论》《新修本草》和《千金翼方》等古代本草书籍均有收载,历史上“桑黄”药名最早出现在《药性论》[1]。现代《中药大辞典》[2]、朝鲜族医药文献《乡药集成方》[3]等有收载。桑黄传统用于止血活血、化饮、止泻,主治脱肛泻血、崩中带下、瘕积聚、利五脏、伏血等症[2]。桑黄是国际高度关注的食药用菌之一,现代研究证明其具有抗氧化活性,能用于治疗癌症,与癌症化疗药物联用具有增效减毒作用,具有提高机体免疫力、保肝护肝、抗菌消炎等功效[4-6]。
桑黄的记载已有两千多年历史,依据古籍对桑黄性状的描述,经过现代分类学家研究发现,外观相似的真菌多种,确定桑黄非单一来源[7]。收载桑黄的法定标准有《湖北省中药材标准》[8]、甘肃省中藏药材标准[9]、《安徽省中药饮片炮制规范》[10]等,但桑黄药材基源各不同。
桑黄为吉林省朝鲜族习用药材,是吉林省药用菌重要栽培品种,长白山地区“特色药材”。该药材法定标准收载于《吉林省中药材标准》[11]和《吉林省中药饮片炮制规范》[12]。本文采用微波消解-电感耦合等离子体质谱法(inductively coupled plasma mass spectrometry,ICP-MS)、薄层色谱法和紫外-可见分光光度法,对不同产地桑黄的安全性与质量进行考察,旨在为桑黄质量标准的建立与评价提供依据。
1 仪器与材料
1.1 仪器
TLC Visualizer 2 薄层色谱数码成像系统(瑞士CAMAG 公司);MARS6 微波消解仪(美国CEM 公司);NexION 2000 电感耦合等离子体质谱仪(美国Perkin Elmer 公司);UV2550 紫外-可见分光光度计(日本岛津公司);TU-1900 紫外-可见分光光度计(北京普析通用仪器有限责任公司);XS105 电子天平(美国梅特勒公司)。
1.2 试剂及试药
硅胶G 薄层板(青岛海洋化工厂分厂);甲苯(国药集团化学试剂有限公司);甲酸(天津市科密欧化学试剂有限公司);硝酸(Fisher Chemical);蒽酮(上海麦克林生化科技有限公司);硫酸(北京化学试剂公司);亚硝酸钠、乙酸乙酯、硝酸铝、氢氧化钠(均为北京化工厂);实验用水均为超纯水。
1.3 样本、对照品及对照药材
25 种元素混合标准溶液(批号:19D6230);汞(Hg,批号:191046-3);铟(In,批号:193014);钪(Sc,批号:193021);铋(Bi,批号:193022-1);金(Au,批号:191095-2)。以上元素标准溶液来源为国家有色金属及电子材料分析测试中心。D-无水葡萄糖(中国食品药品检定研究院,批号:110833-201707,含量:99.9%);芦丁(中国食品药品检定研究院,批号:100080-201811,含量:91.7%);桑黄对照药材[由海南医学院范宇光副教授鉴定,基原为锈革孔菌科瓦宁木层孔菌Sanghuangporus vaninii(Ljub) L.W.Zhou &Y.C.Dai[11]]。
样本来源见表1。

表1 样本来源
2 方法与结果
2.1 方法
2.1.1 薄层色谱法(thin layer chromatography,TLC)鉴别方法
取样本粗粉1.5 g,加甲醇60 ml,超声30 min,滤过,滤液蒸干,残渣加甲醇3 ml 使溶解,作为供试品溶液。另取桑黄对照药材1.5 g,同法制成桑黄对照药材溶液。按照薄层色谱法试验[13]59,吸取样本与桑黄对照药材溶液各5 μl,分别点于同一硅胶G 薄层板上,以甲苯-乙酸乙酯-甲酸(8∶5∶2)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。
2.1.2 微波消解-ICP-MS 测定法
2.1.2.1 对照品的制备
将混合标准溶液用(5%+95%)的硝酸依次按1∶50∶1.667∶3∶2∶5 依次稀释成相应浓度的标准系列溶液,将汞单元素标准溶液用(5%+95%)的硝酸配制成0.1、0.5、1、2、3 μg/L 系列标准溶液,现配现用,将In、Bi、Sc 配制成浓度为100 μg/L 的内标混合溶液。
2.1.2.2 样本的处理
称取样品粗粉0.5 g,置微波消解内罐中,加入7 ml硝酸,用微波消解法消解完全,加入200 μl 浓度为1 μg/ml 的Au 单元素标准溶液,将消解罐放入石墨消解仪上100℃加热30 min,用水定容至50 ml,混匀备用。
除不加Au 单元素标准溶液外,同法制备试剂空白。
2.1.2.3 加样回收率
称取2 号样品6 份,每份0.5 g,分别加入26 种元素标准品,然后按样品操作,测定加样回收率。
2.1.2.4 实验条件
消解仪条件:5 min 由室温升到100℃保持3 min,7 min 由100℃升到150℃保持10 min,7 min 由150℃升到180℃保持20 min。
ICP-MS 条件:射频发生器功率为1600 W,碰撞气为氦气,载气为氩气,雾化气流量为1.04 L/min,驻留时间为50 ms,雾化室温度为2℃,数据采集重复次数为3 次。
2.1.3 桑黄多糖含量测定方法
2.1.3.1 样本的处理
取样本粗粉0.9 g,精密称定,置锥形瓶中,加水100 ml 使样品充分润湿,静置1 h,加热回流4 h,趁热滤过,用少量热水洗涤滤器和滤渣,将滤渣及滤纸置锥形瓶中,加水100 ml,加热回流3 h,趁热滤过,用少量热水洗涤滤器和滤渣,合并滤液,置水浴上蒸干,残渣用水10 ml 溶解,边搅拌边缓慢滴加无水乙醇90 ml,摇匀,在4℃放置12 h,离心,弃去上清液,沉淀物用热水溶解并转移至25 ml 量瓶中,放冷,加水至刻度,摇匀,取溶液适量,离心,精密量取上清液5 ml 置50 ml 量瓶中,加水至刻度,摇匀,即得。测定法参照《中华人民共和国药典》(2020年版·一部)灵芝项下[14]195。
2.1.3.2 方法学
2.1.3.2.1 线性与范围 精密称取于105℃干燥至恒重的无水葡萄糖对照品12.01 mg,置100 ml 容量瓶中,加水制成每1 ml 含0.1201 mg 的溶液。标准曲线制备参照《中华人民共和国药典》(2020年版·一部)灵芝项下方法[14]195进行配制并测定,绘制标准曲线。
2.1.3.2.2 精密度试验 ①重复性:取5 号样品粗粉0.45 g,精密称定,置具塞锥形瓶中,共9 份,对照品按1.5∶1、1∶1、0.5∶1 加入,每个比例制备3 份,按样本处理方法提取测定;②中间精密度:取5 号样品粗粉0.9 g,不同操作人员,不同的紫外-可见分光光度计,不同检测日期,按样本处理方法提取测定,测定桑黄多糖含量。
2.1.3.2.3 准确度试验 取已知含量5 号样品粗粉0.45 g,精密称定,置具塞锥形瓶中,共9 份,对照品按1.5∶1、1∶1、0.5∶1 加入,每个比例制备3 份,按样本处理方法提取测定,计算加样回收率。
2.1.3.2.4 耐用性试验 ①紫外-可见分光光度计考察:分别采用二个不同品牌的紫外可见分光光度计(岛津UV2550,带宽分别为0.5、1、2 nm,普析通用TU-1900,带宽为2 nm);②试剂考察:分别采用不同厂家的蒽酮和硫酸,配制硫酸蒽酮溶液,测定同一样品中多糖的含量。
2.1.4 桑黄总黄酮含量测定方法
2.1.4.1 样本的制备
取本品粗粉约0.3 g,精密称定,加60%乙醇60 ml,加热回流提取2 h,放冷,滤过,用60%乙醇35 ml,分次洗涤残渣,合并滤液,置100 ml 量瓶中,用60%乙醇稀释至刻度,摇匀,即得。
2.1.4.2 对照品溶液的制备精密称取芦丁对照品适量,加60%乙醇适量,制成每l ml 中含芦丁0.2 mg 的溶液,即得。
2.1.4.3 标准曲线的制备
精密量取对照品溶液l、2、3、4、5、6 ml,分别置25 ml 量瓶中,各加水至6 ml,加5%亚硝酸钠溶液1 ml,混匀,放置6 min,加10%硝酸铝溶液l ml,摇匀,放置6 min,加氢氧化钠试液10 ml,再加水至刻度,摇匀,放置15 min。以相应的试剂为空白,通过全波长扫描,确定波长为509 nm,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
2.1.4.4 精密度试验
①重复性:取5 号样品粗粉0.15 g,精密称定,置具塞锥形瓶中,共9 份,对照品按1.5∶1、1∶1、0.5∶1 加入,每个比例制备3 份,按样本制备方法提取、测定。②中间精密度:取5 号样品粗粉0.3 g,不同操作人员,不同紫外-可见分光光度计,不同检测日期,按样本制备方法提取,测定。
2.1.4.5 准确度试验
取已知含量5 号样品粗粉0.15 g,精密称定,置具塞锥形瓶中,共9 份,对照品按1.5∶1、1∶1、0.5∶1 加入,每个比例制备3 份,按样本制备方法提取、测定,计算加样回收率。
2.1.4.6 耐用性试验
紫外-可见分光光度计考察:分别采用二个不同品牌的紫外-可见分光光度计(岛津UV2550,带宽分别为0.5、1、2 nm,普析通用TU-1900,带宽为2 nm),测定样品中总黄酮的含量。
2.2 结果
2.2.1 TLC 鉴别结果
进行耐用性和不同产地样本考察,结果表明,TLC 鉴别的耐用性良好,专属性强,10 批不同产地的样本薄层色谱图见图1,结果显示,其薄层色谱图基本一致,此方法的建立能有效保证桑黄的质量。

图1 TLC 色谱图
2.2.2 ICP-MS 测定结果
26 种元素的标准曲线、r值、线性范围等参数见表2。相应浓度范围内回归方程r值均高于0.995,线性关系良好。平行制备11 个试剂空白,测定试剂空白值,检出限为3 SD。同一样本重复测定7 次,计算仪器精密度,结果良好。加样回收率和r值均符合《中华人民共和国药典》(2020年版·四部)规定[13]482。
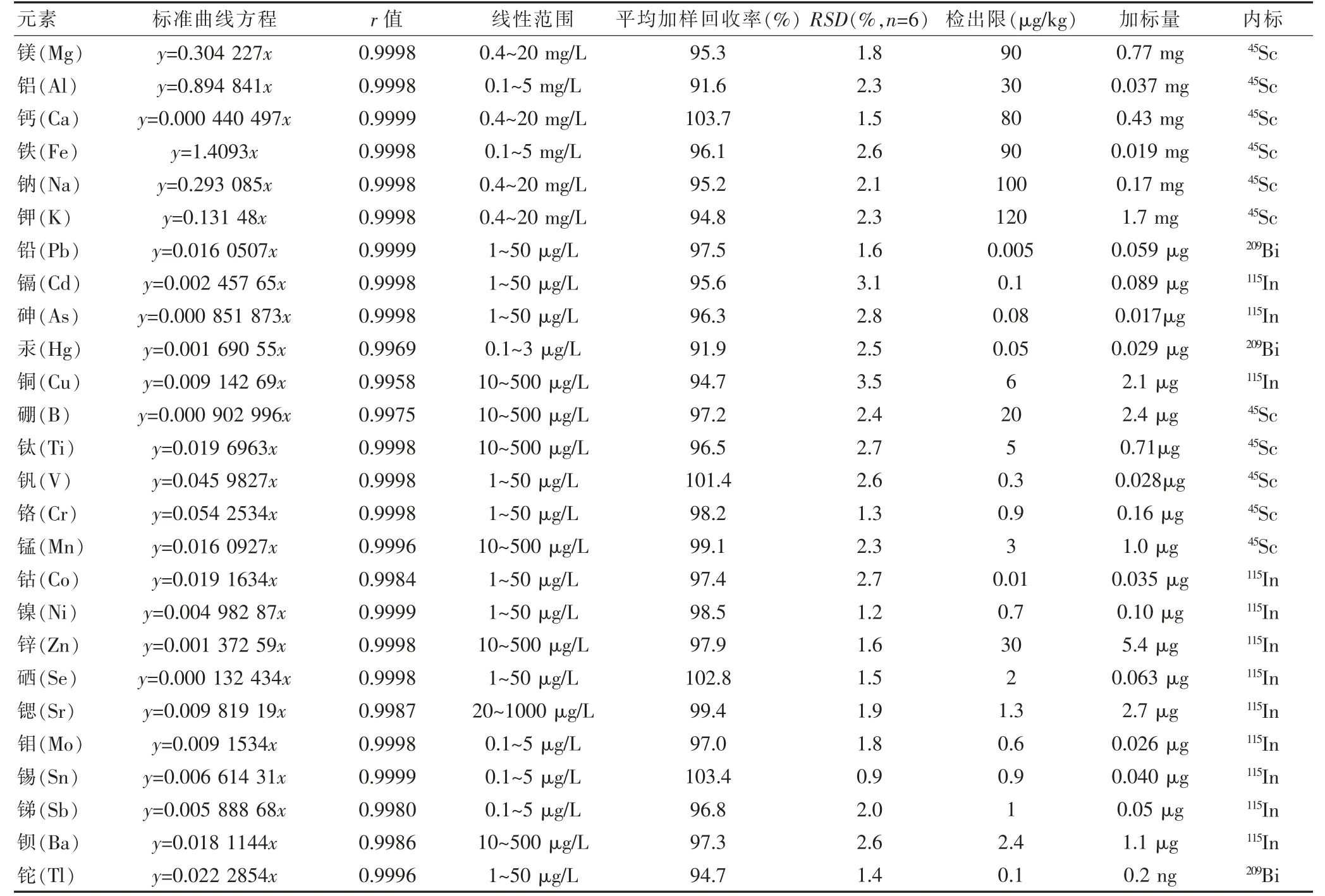
表2 桑黄的ICP-MS 方法学参数
10 批桑黄样本的各元素含量检测结果见表3。
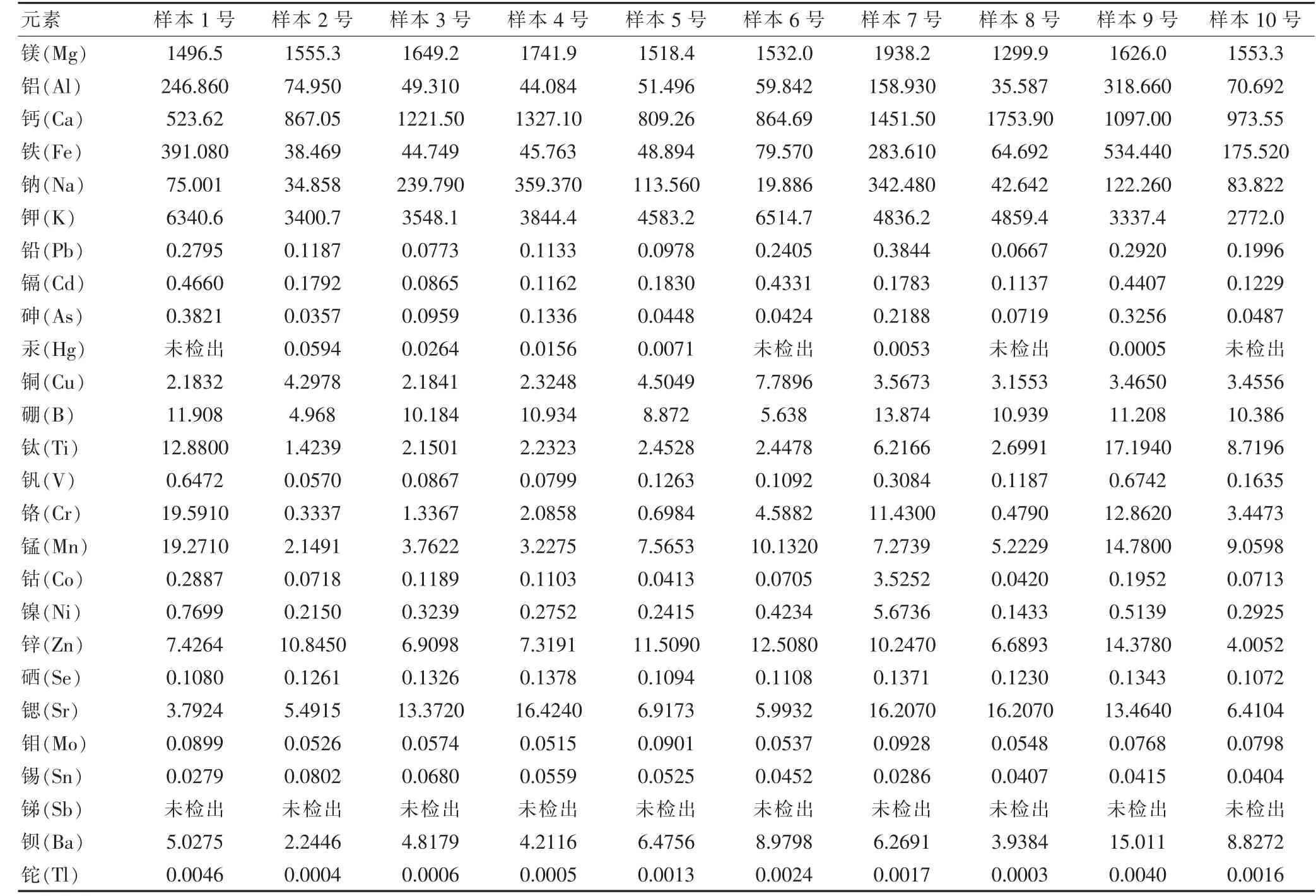
表3 桑黄的ICP-MS 检验结果(mg/kg)
2.2.3 桑黄多糖含量测定结果
2.2.3.1 方法学
2.2.3.1.1 线性与范围 无水葡萄糖在0.024~0.120 mg范围内与吸光度呈良好的线性关系。回归方程为:y=0.937 95x+0.133 29,r=0.9982。
2.2.3.1.2 精密度试验 ①重复性:结果平均值为1.43%,RSD=1.6%(n=9),表明该法的重复性良好。②中间精密度:结果平均值为1.42%,RSD=2.9%(n=6)。表明该法的中间精密度良好。
2.2.3.1.3 准确度试验 平均回收率为94.6%,RSD=1.5%(n=9)。
2.2.3.1.4 耐用性试验 ①紫外-可见分光光度计考察:结果显示,不同仪器和带宽对实验结果的影响,无明显差异;②不同试剂的考察:6 种组合配制成的硫酸蒽酮溶液的测定结果平均值为1.44%,RSD=1.8%(n=6)。
2.2.3.2 样本含量
10 批桑黄多糖含量测定结果见表4。
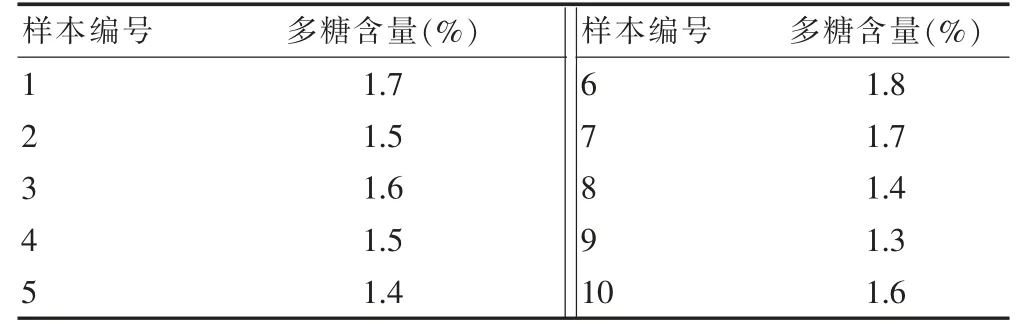
表4 多糖含量测定结果
2.2.4 桑黄总黄酮测定结果
2.2.4.1 方法学
2.2.4.1.1 线性及范围 总黄酮以芦丁计在0.2~1.2 mg范围内与吸光度呈良好的线性关系。回归方程为:y=0.473 61x-0.0112,r=0.9996。
2.2.4.1.2 精密度试验 重复性测定结果平均值为8.77%,RSD=1.4%(n=9),试验表明该法的重复性良好;中间精密度含量测定结果平均值为8.76%,RSD=2.7%(n=6),结果表明该法的中间精密度良好。
2.2.4.1.3 准确度试验 总黄酮平均回收率为96.3%,RSD=1.4%(n=9)。
2.2.4.1.4 耐用性试验 分别采用两个不同品牌的紫外-可见分光光度计测定,测定结果平均值为8.74%,RSD=1.8%(n=4),显示不同仪器和带宽对实验结果的影响,无明显差异。
2.2.4.2 样本含量10 批桑黄总黄酮的含量测定结果见表5。
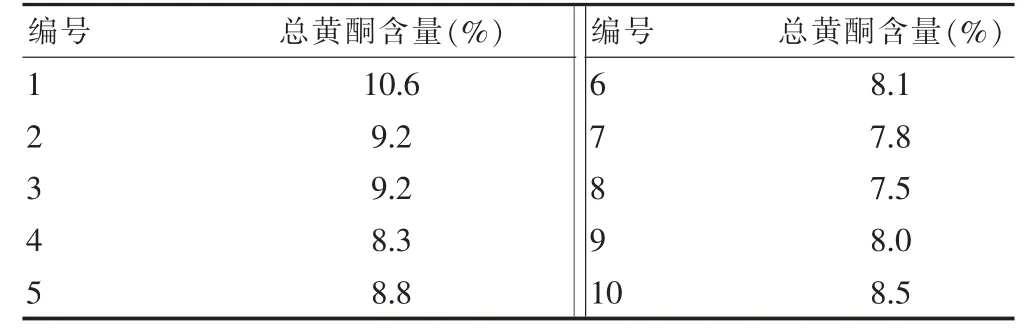
表5 总黄酮含量测定结果
3 讨论
对薄层色谱鉴别中提取溶剂(甲醇、乙酸乙酯、石油醚)及提取方法(回流提取与超声处理)进行了考察。结果表明提取溶剂为甲醇时,提取成分较多,同时回流提取与超声处理无明显差别,由于超声方法处理简单,重复性与稳定性良好,易于检验,故采用甲醇超声作为桑黄薄层鉴别处理方法。
通过微波消解-电感耦合等离子体质谱法测定10 批不同产地桑黄中26 种元素的含量。结果分析:①铅、镉、砷、汞、铜为《中华人民共和国药典》(2020年版·四部)规定的5 种重金属及有害元素,参照人参项下相关规定[14]8,桑黄样本均未超标。②高含量的5 种有益元素,分别为钾(2772.0~6514.7 mg/kg)、镁(1299.9~1938.2 mg/kg)、钙(523.62~1753.90 mg/kg)、钠(19.886~359.370 mg/kg)和铁(38.469~534.440 mg/kg)。钾元素能维持水分平衡、调节血流量、影响神经传导和对肌肉正常收缩协助等生理作用,对治疗跌打损伤和活血理气有辅助疗效[15]。其中钾元素含量最高,与桑黄的活血、化饮、治瘕积聚、利五脏疗效相符。铁元素参与细胞色素的合成、氧的运输和贮存及提高机体免疫,可能与桑黄提高机体免疫力相关。据报道大运动量的极限或亚极限的力竭训练后,人体钾、镁、钙、钠等元素的流失,会引起运动型心源疲劳[16]。桑黄中钾、钙、钠、镁元素含量高,对其开发改善心脏功能的产品,有重要意义。③锑为高致癌元素[17]、铊元素和钡元素有人体中毒剂量[18-19],锑元素未检出,铊元素、钡元素含量低,均未超标。锌和硒元素含量较低,剩余11种元素,按每日使用最大剂量30 g[2]计算,摄入量低,同时无桑黄人体中毒报道。26 种元素的检测,目前为止未发现安全风险,为桑黄安全性提供依据。
研究证明,桑黄多糖具有抗肿瘤、改善机体有氧运动、抗氧化和保护造血机能损伤等作用[20-23],桑黄的总黄酮具有抗癌和抗氧化活性[24-25],因此建立桑黄多糖与总黄酮检验方法。
本研究同时对10 批不同产地桑黄进行考察,结果重复性与稳定性良好,易于检验,能够为桑黄质量标准的研究与开发提供依据。
