煤族组分骨架结构的分子模型构建及分子动力学模拟
2021-10-26连露露秦志宏李春生杨小芹
连露露,秦志宏,李春生,杨小芹,林 喆
(中国矿业大学 化工学院,江苏 徐州 221116)
从分子水平上认识煤结构、煤反应性及煤形成过程中的变化规律是煤科学研究的热点之一。众多学者基于原煤的元素组成和结构参数构建了相应的煤结构模型[1-9],但考虑到煤组成和煤结构的高度复杂性,这种建立在原煤基础上的煤结构模型明显粗糙,只能反映原煤这种广泛意义上的结构平均,而不能细致反映出煤中不同族组分的结构特点及差别,其代表性明显不足。
笔者[10]提出认知煤结构应采用“模糊/精细论”的思想方法,即利用全组分分类法对煤这一复杂体系进行“种属”划分,然后各个击破,再分别对仍然复杂的各种“属”采用模糊方法进行较细致的解析,最后统一到煤组成结构的大系统中,从而在宏观层面解决对煤组成结构的系统认知难题,并由此创立了煤嵌布结构模型理论。通过对煤的CS2/NMP混合溶剂萃取液进行多次反萃取操作,将煤在常压室温下分离成了组成、结构和性质完全不同的4种族组分,指出煤是由这些族组分以物理尺度为100 nm左右的颗粒互相嵌布而成,这些颗粒包括疏中质组(LMC),密中质组(DMC)和重质组(HC)3类族组分,而第4类族组分轻质组(LC)则以小分子方式在这些颗粒间起着桥联作用。每种颗粒型族组分都由骨架部分和小分子部分所组成;其中骨架部分由若干个大分子聚集形成,而赋存型有机小分子充填在这些骨架结构中。进一步研究发现,不同的族组分颗粒不仅在组成结构上存在显著差异,而且还表现出完全不同的反应性能。例如在炼焦煤热解生成胶质体的过程中,DMC是专门产生流动性的组分,而LMC则是专门产生膨胀性的组分[11]。另外,DMC还是煤热解生成中间相小球体的来源物质[12]。
以分离出的各类族组分为基础的煤嵌布结构模型理论,展现了对煤结构认知的系统性和全面性特点。根据该理论,由分子尺度进一步弄清各族组分结构细节及其形成的分子机制,无疑将深化对煤结构的分子认知,为研究煤的多组成性及其之间的相互作用提供基础,并对煤加工转化利用技术开发提供借鉴。
在计算技术迅速发展的今天,分子力学及分子动力学方法己成为构建三维煤结构模型并从分子尺度研究煤的结构与性质的常用方法[13-14],例如,JONES等[2]创建了煤3D结构模型并采用分子力学方法计算了煤结构体系的键能和非键能;LIU等[15]采用分子力学方法研究了亚烟煤镜质组中的超微孔结构特征;ZHANG[16]利用分子动力学模拟了褐煤除湿过程中基体的结构构象变化。分子力学及分子动力学模拟在煤表面结构和性质研究中获得了更多的应用[17-19],但用于煤中大分子间相互作用及形成聚集体的分子机制研究鲜见报道。
笔者拟采用分子力学及分子动力学方法,在煤嵌布结构模型理论基础上,进一步探究煤中各族组分骨架大分子形成骨架网络结构的分子机制和能量机制。主要完成以下目标:① 构建各族组分骨架分子结构模型;② 探究各族组分骨架分子形成大分子网络结构的分子机制;③ 从分子层面,解析煤中各族组分骨架分子稳定态及多分子稳定聚集体形成机制。
1 实验方法
1.1 煤族组分分离
选用淮北童亭原煤(Raw coal)为实验用煤,将其磨碎至0.074 mm(200目)后密封保存。采用萃取反萃取方法分离得到HC,LMC,DMC和LC四大族组分。制备流程及各族组分产率(质量分数,daf)如图1所示(实验中有4.37%的样品损失)。各族组分分别干燥称重后密封保存。原煤及各族组分的工业分析与元素分析见表1。
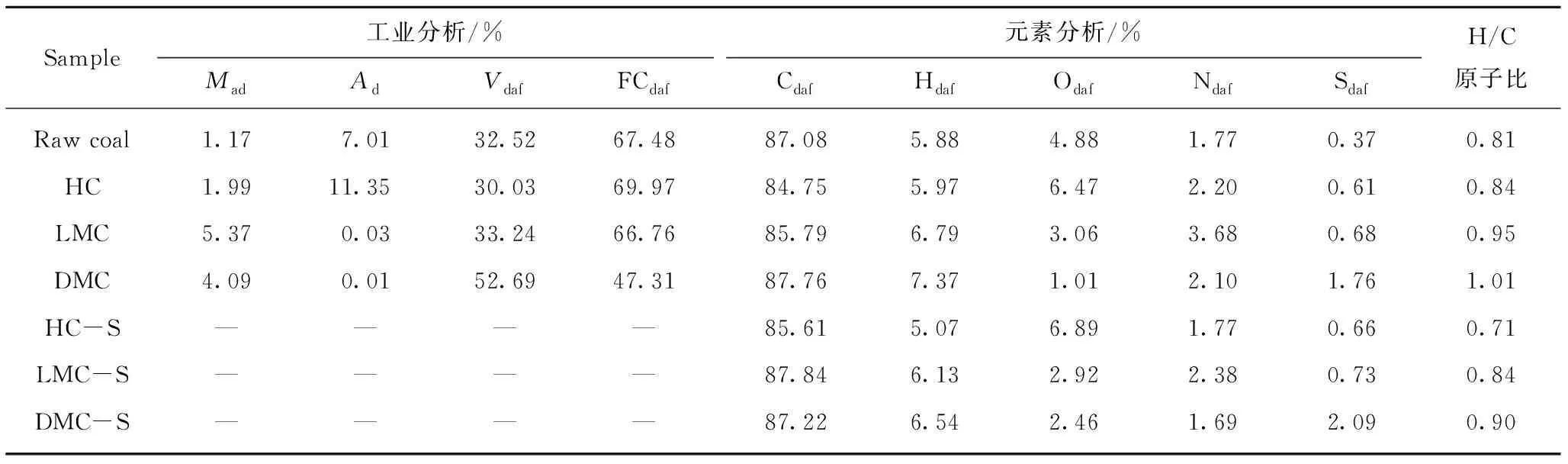
表1 原煤、族组分及族组分骨架的工业分析和元素分析Table 1 Element analysis of raw coal,all group component,and group component skeleton
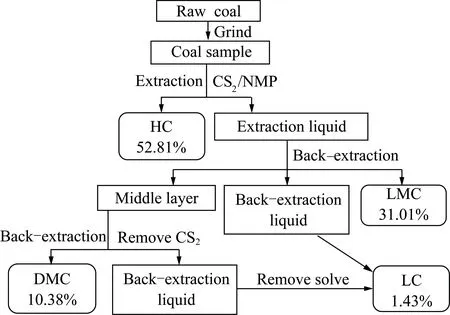
图1 煤全组分族分离流程及各族组分产率Fig.1 Flow diagram of coal all-group separation process and the yields of group component
1.2 族组分骨架的获取方法
称取一定量的HC(LMC,DMC)组分,用450 nm聚四氟乙烯膜和滤纸包裹,放入22 mL快速溶剂萃取仪的萃取池中,依次用正己烷、甲醇、丙酮和氯仿溶剂进行分级分次静态萃取(其中正己烷萃取对HC 4次,对LMC 5次,对DMC 7次;甲醇萃取对HC 3次,对LMC 4次,对DMC 4次;丙酮萃取对HC 6次,对LMC 6次,对DMC 5次;氯仿萃取对HC 8次,对LMC 5次,对DMC 4次。各级的萃取次数依据最后一次萃取液是否接近无色且对其进行GC/MS检测时已无出峰或出峰丰度很低来综合判定),流程如图2所示。每次萃取均控制萃取温度100 ℃,压力10 MPa,萃取时间10 min。萃取结束后,用旋转蒸发仪除去萃取液中大部分溶剂后,进行GC/MS检测;真空干燥后的萃余物即为该族组分的骨架部分,用HC-S(LMC-S,DMC-S)表示。LC产率较低,主要是有机小分子,在4级萃取中完全溶解至萃取液中,不存在骨架结构的概念。
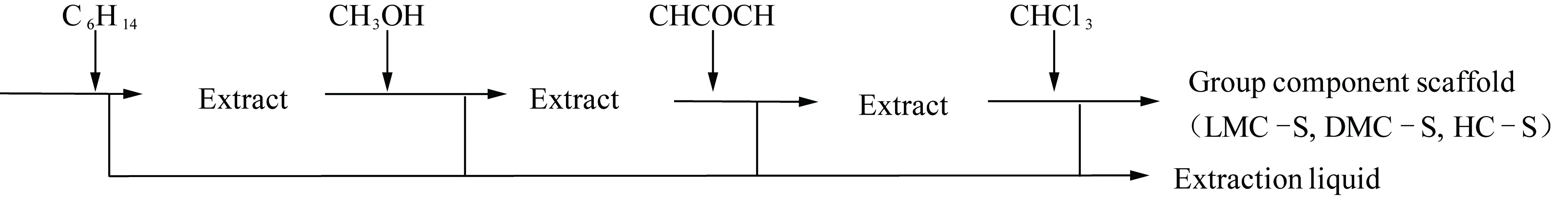
图2 各族组分分级分次萃取流程Fig.2 Fractional extraction process of group components
4种族组分溶出物中均含有杂原子化合物及脂肪族化合物,其中HC溶出物中杂原子化合物种类和数量均比LMC,DMC多;LC组分溶出物以含N和O 的杂原子化合物为主,分子量在85~200;DMC组分溶出物中包括有C10~C24正构烷烃、支链烷烃和带有取代基的环己烷、环戊烷,其中环烷烃的种类较其他族组分明显偏多。HC,LMC及DMC组分溶出物中均有芳香类化合物,LC组分没有检测到芳香类化合物。
1.3 族组分骨架的结构分析
1.3.1元素分析
采用Elementar vario Macro全自动元素分析仪测定各族组分骨架中的元素含量,结果见表1。
1.3.2FTIR分析
采用Thermo Nicolet 公司生产的傅里叶变换红外光谱分析仪进行表征,样品经研磨、烘干后通过KBr压片法制样,煤样与KBr的质量比为1∶100。样品扫描 16次,光谱范围 400~4 000 cm-1。
1.3.3XPS分析
采用ESCAlab 250Xi 型 X射线光电子能谱仪进行表征,单色化Al Ka阳极作为激发源。全扫透过能为100.0 eV,步长1.0 eV;固定窄扫透过能20.0 eV,步长 0.050 eV。以C1S(284.8 eV)做内标进行校正。
1.3.413C NMR分析
采用Bruker Avance Ⅲ 300 MHz超导核磁共振仪进行表征,利用交叉极化和魔角旋转技术,固体双共振探头,4 mm WVT转子,转速为5 000 Hz,13C检测核的共振频率为75.48 MHz,谱宽500×10-6,采样时间为18 ms,循环延迟时间2 s,碳氢交叉极化接触时间1 000 μs,扫描次数为2 048,以金刚烷为定标化合物。
1.4 构建并校正各族组分骨架分子结构模型
在ChemDraw软件中绘制族组分结构模型并用Gaussian 09W软件对构建的结构模型进行频率计算,结果与实验红外谱图对比并不断调整分子结构模型,最终确定各族组分骨架分子结构模型。
1.5 TEM分析
采用JEM-200CX型透射电子显微镜对CS2/NMP混合溶剂萃取液进行测试。测试过程中用专用的铜网置于该萃取液中,缓慢移动约30 s后取出铜网,在真空干燥箱进行24 h真空干燥后进行TEM观察。
2 模拟方法
2.1 族组分骨架分子单分子优化
所有模拟均采用Universal[20]力场,总势能计算公式:ETotal=EB+EA+ET+EI+Evdw+Eel。其中键角能(EA)、二面角扭转能(ET)和反转能(EI)构成角能;角能和键键伸缩能(EB)构成价键能(EV);范德华能(Evdw)和静电能(Eel)构成非键能(EN)。
首先对各族组分骨架分子进行单分子几何优化。参数设置如下:总步数为100 000;收敛标准为超精细;能量差为0.004 2 kJ/mol。平衡电荷采用Gasteiger[21]方法,库仑力和范德华力计算采用Atom based。为了消除分子结构的能量脊使得分子处于最优的几何状态,再采用退火模块对分子进行动力学分析。参数设置如下:总步数为4 000 000;收敛标准为超精细;库仑力和范德华力计算采用Atom based。初始温度设置为300 K,终温600 K,升温速率为3 K/次,每个温度都在NVT(恒原子数恒温恒体积)系综下进行动力学模拟,时长1 fs,温度采用Nose[22]法控制,循环次数为20。选取势能最低构型进行下一步分子动力学模拟。
2.2 族组分骨架分子的密度模拟
不同族组分骨架分子的模拟密度方法见文献[23],采用MS 8.0[24]软件Amorphous Cell模块给予分子模型周期性边界条件,然后在不同的周期性边界条件下进行退火动力学分析,不断调整晶胞大小优化结构,根据密度-势能曲线图得出分子最优密度。
2.3 不同配置单元的分子动力学模拟
采用Forcite中Amorphous Cell模块,将已优化的单分子按不同个数并添加周期性边界条件后构成配置单元(1个疏中质组骨架分子构成的配置单元用LMC-S-1表示,2个疏中质组骨架分子构成的配置单元用LMC-S-2表示,以此类推),然后再利用Anneal和Dynamical模块对已构建的配置单元进行动力学分析。初始温度设置为300 K,终温设为600 K,升温速率为3 K/次,在每1个温度下在NVT系综下进行动力学模拟,循环次数为20。总退火动力学模拟时长为4 000 ps。选择势能最低结构模型,再以同样的升温速率在0.01 GPa压力下将系统温度从300 K升温至600 K,最后在NVT系综下进行动力学平衡模拟,平衡动力学模拟时长为5 000 ps,模拟过程中采用Nose’s 恒温器[22]和Berendsen’s 恒压器[25]控制温度和压力。
3 结果与讨论
3.1 各族组分中元素存在形态
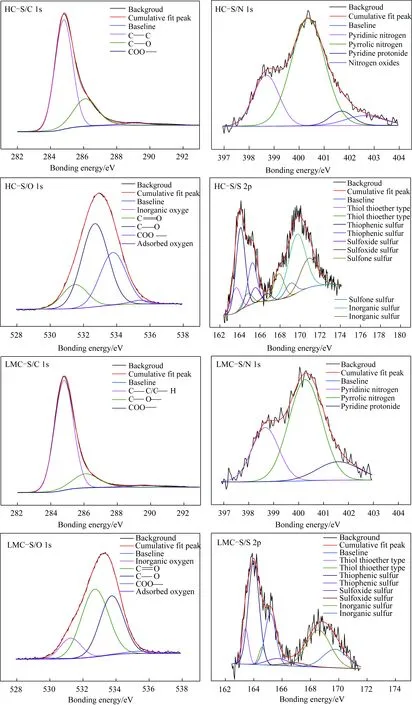
本文中C(1s)、O(1s)、N(1s)及S(2p)的XPS吸收峰均由XPSPEAK4.12软件进行分峰拟合,选择Lorentzian-Gaussian混合峰形(图3),XPS吸收峰归属参见文献[26-27],结果见表2。由于矿物中氧含量影响O1s谱图,因此煤中碳和氧形成的键以C1S分析数据为准。

表2 族组分骨架的C(1s),O(1s),N(1s)和S(2p)XPS分析数据Table 2 XPS C(1s),O(1s),N(1s)and S(2p) data of group components
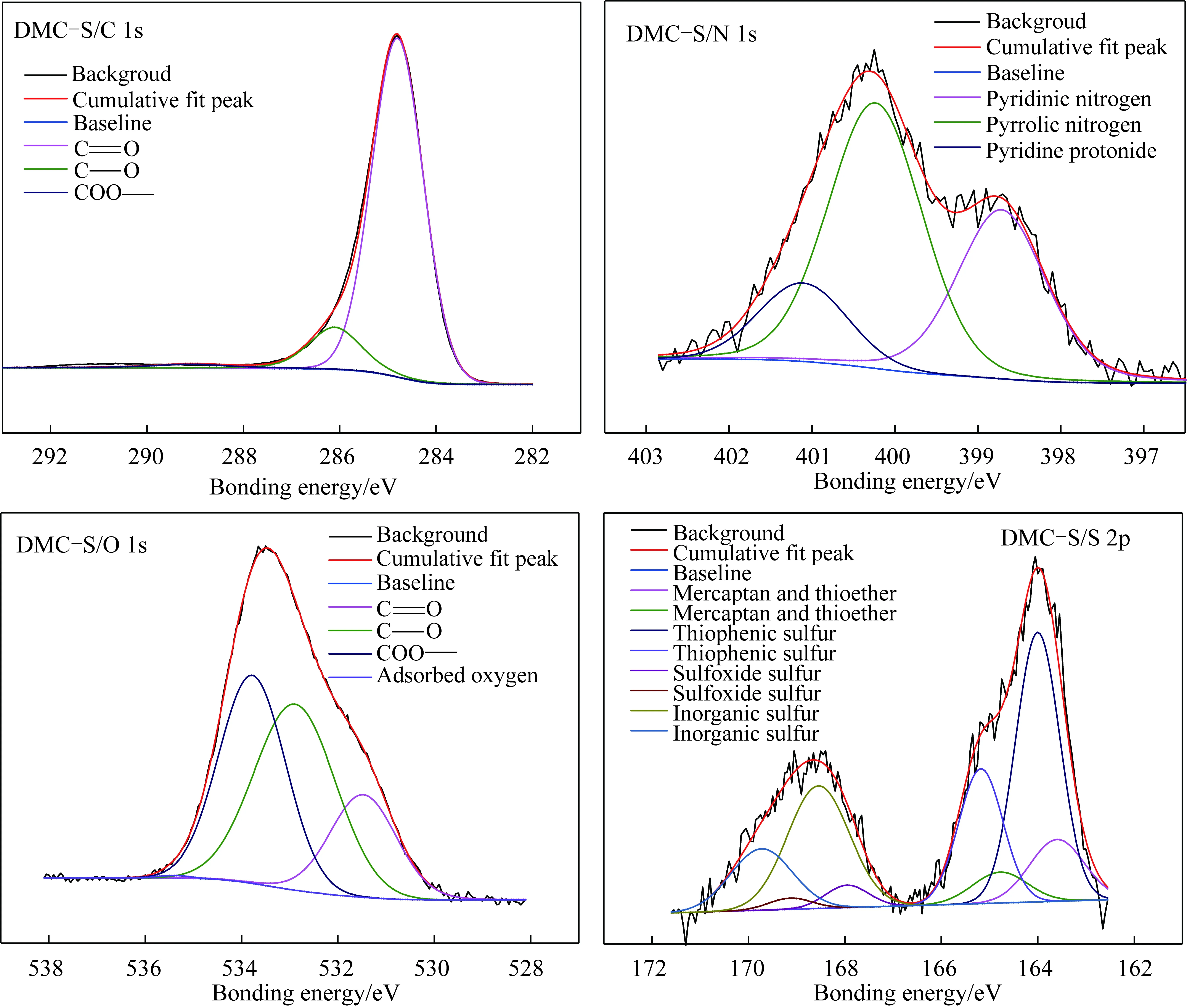
图3 族组分骨架的C(1s),O(1s),N(1s)和 S(2p)XPS分析Fig.3 XPS C1s spectrum,XPS O1s spectrum,XPS N1s spectrum,XPS S2p spectrum of group components skeletons
3.2 族组分骨架分子的结构参数
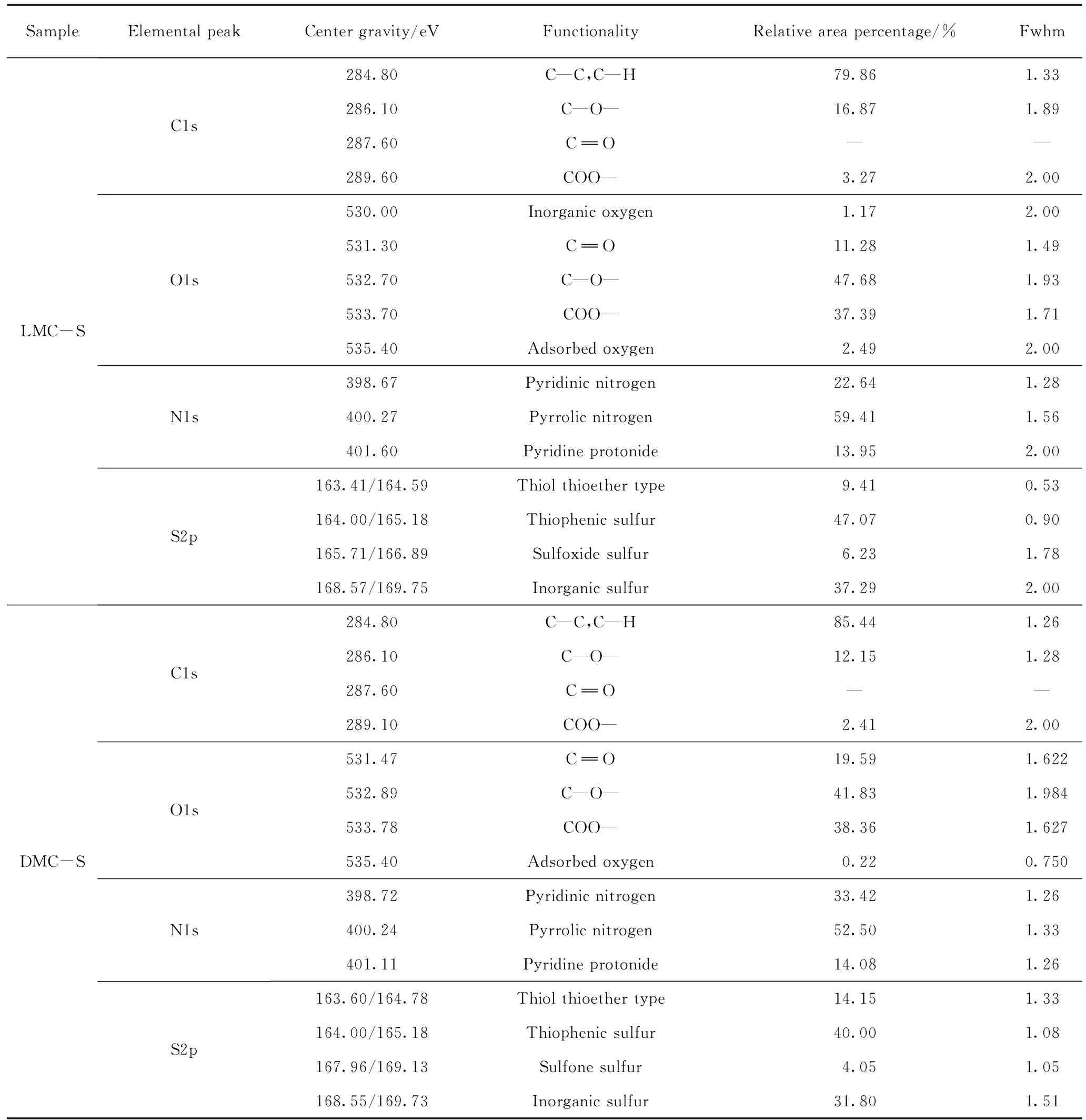
依据13C NMR 检测结果,采用NUTS2000软件进行分峰拟合,根据化学位移并参考相关文献获得碳原子的归属[28-29],由分峰拟合结果获得相应的积分相对含量,在此基础上计算得出各族组分骨架的12个分子结构参数,结果见表3。
续 表
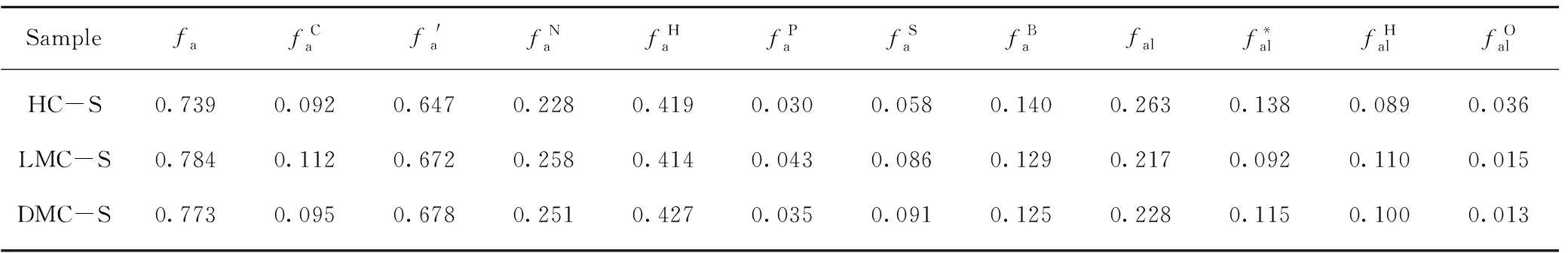
表3 族组分骨架的分子结构参数Table 3 Structural parameters of group components skeletons
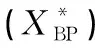
表4 族组分的原子数量及芳香桥碳和周碳之比的实验值(XBP)、计算值Table 4 Calculation and experimental values of aromatic bridgehead to surrounding and the atomic number of group components
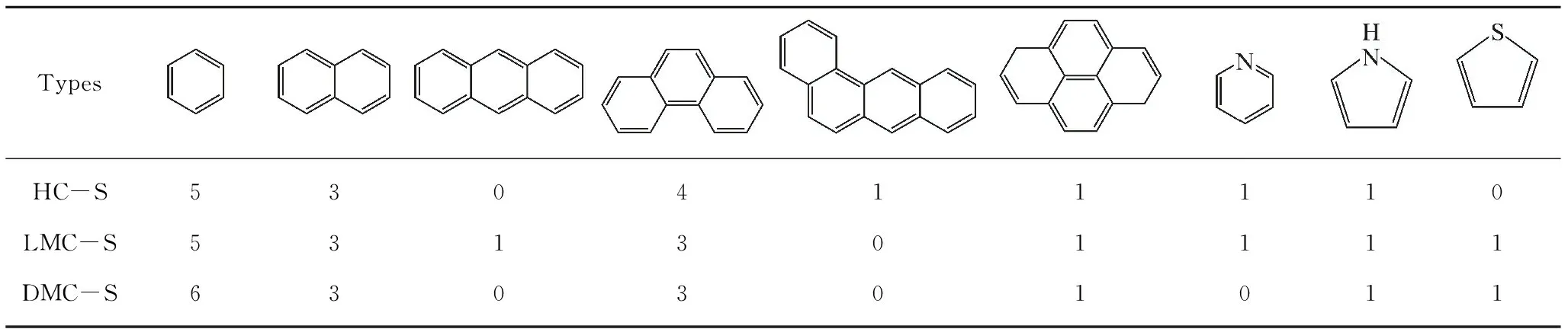
表5 族组分骨架的芳香结构单元数量Table 5 Types of aromatic structure units in the chemical structural model of group components

3.3 族组分骨架分子结构模型构建及验证
采用ChemDraw软件初步绘制出各族组分骨架部分的分子模型,然后应用Gaussian 09W软件,在HF/3-21G水平上完成初步的能量与频率计算,所得IR图谱与实验FTIR谱图有较好的对应关系为止(图4)。最终确定各族组分的分子结构模型如图5所示。
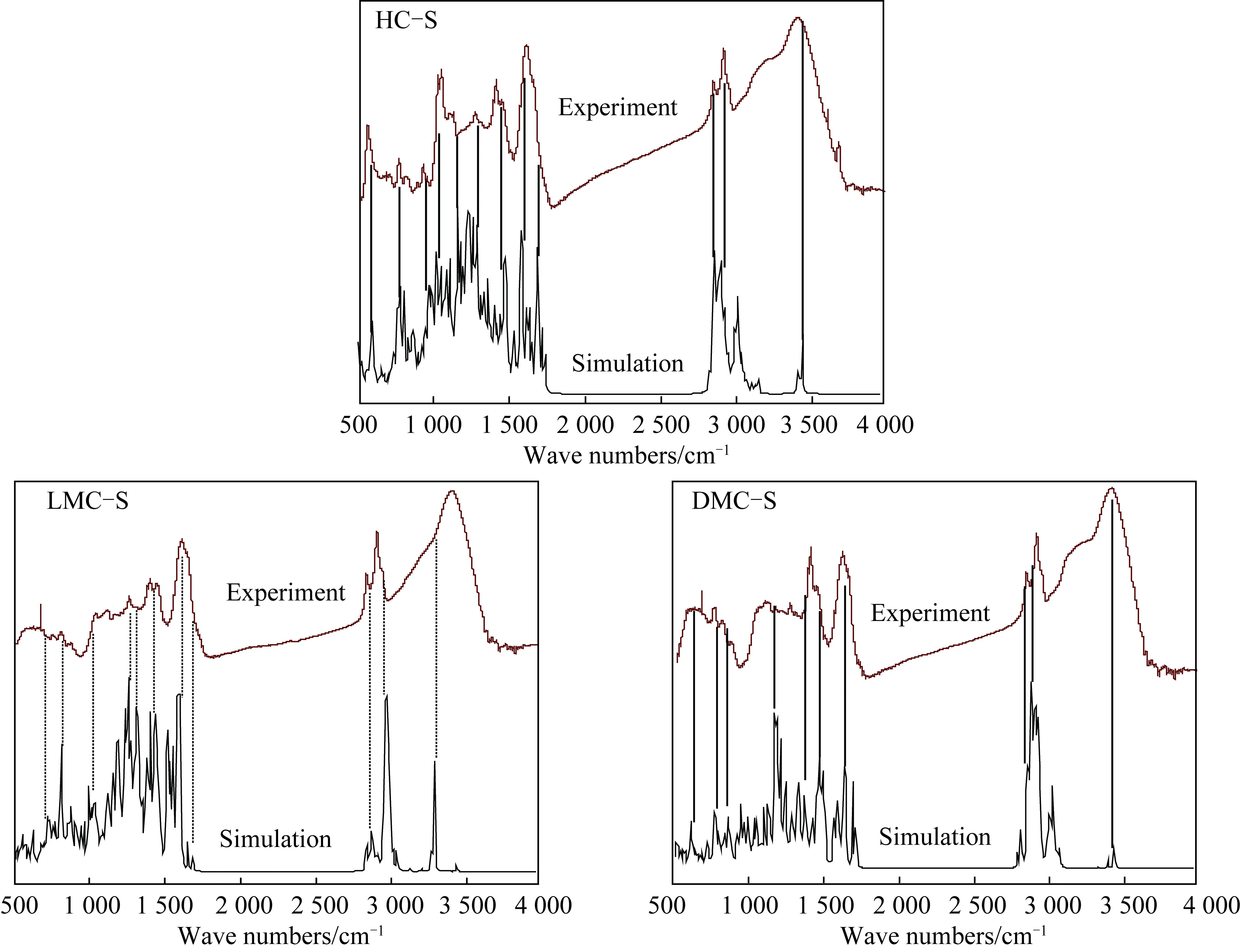
图4 族组分骨架的IR谱图与实验FTIR谱图对应关系Fig.4 Comparing calculated and experimental spectra of group components skeletons
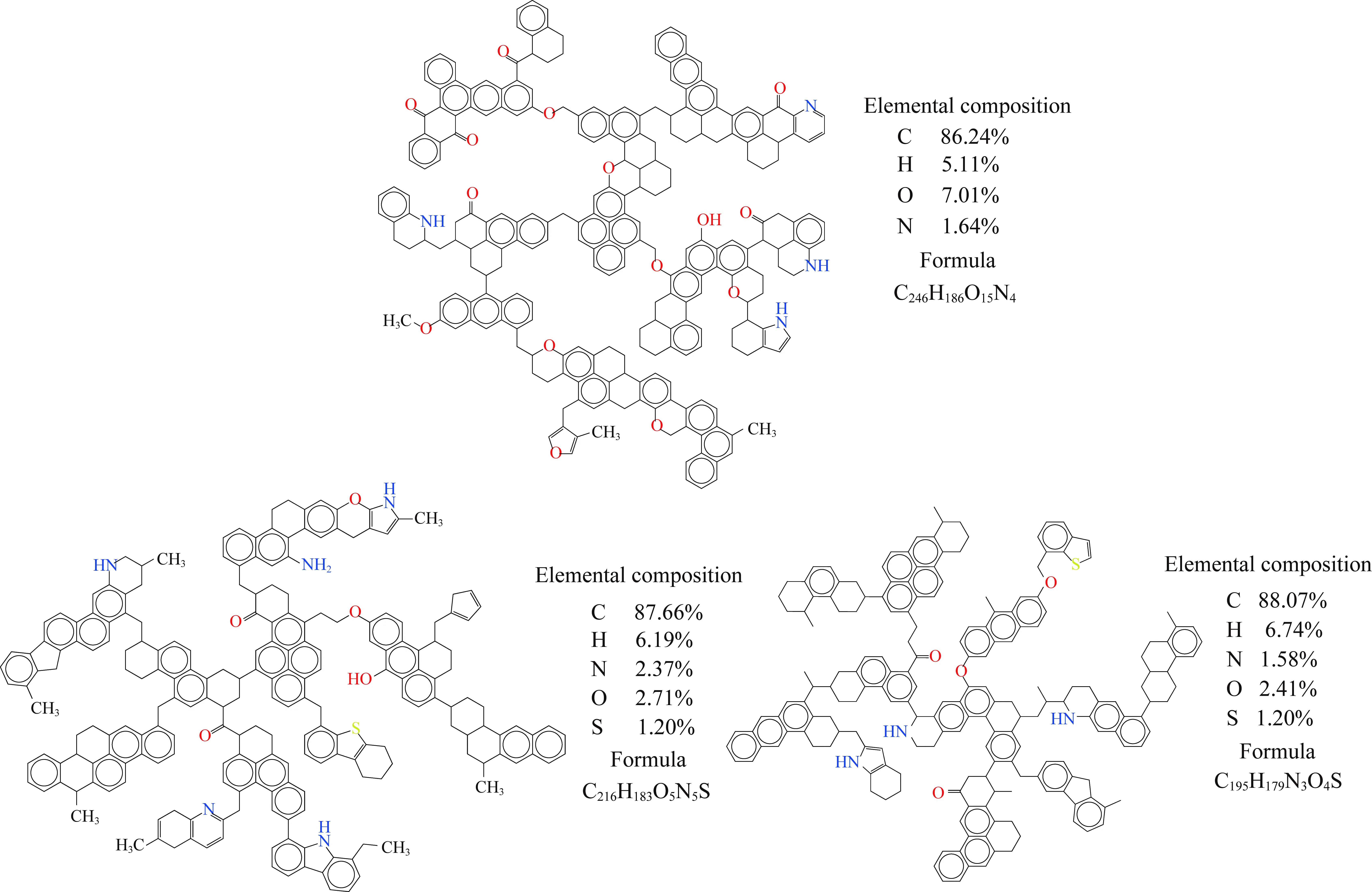
图5 构建的族组分骨架分子结构模型Fig.5 Chemical structural model of group component skeleton
3.4 各族组分单个骨架分子结构的优化
对构建的各族组分分子结构模型进行几何优化及退火动力学优化后,得到的势能最低构型如图6所示。DMC-S 结构中含有较多的单键,且支链中的官能团含环状结构数量较少,单键较易发生扭转;其芳香环部分几乎以平行的方式排列,易形成 π-π 共轭作用,因此其势能最低构型接近球型且非常紧凑。与此相反,HC-S结构中单键较少,且支链中的官能团含环状结构数量较多,易形成π-π及π-σ超共轭作用,因而其构型呈层片状且较为松散。LMC-S 最低势能构型的紧凑程度位于DMC-S和HC-S之间。HC-S总势能最大,LMC-S次之,DMC-S最小。不同族组分的能量参数也不同(表6)。
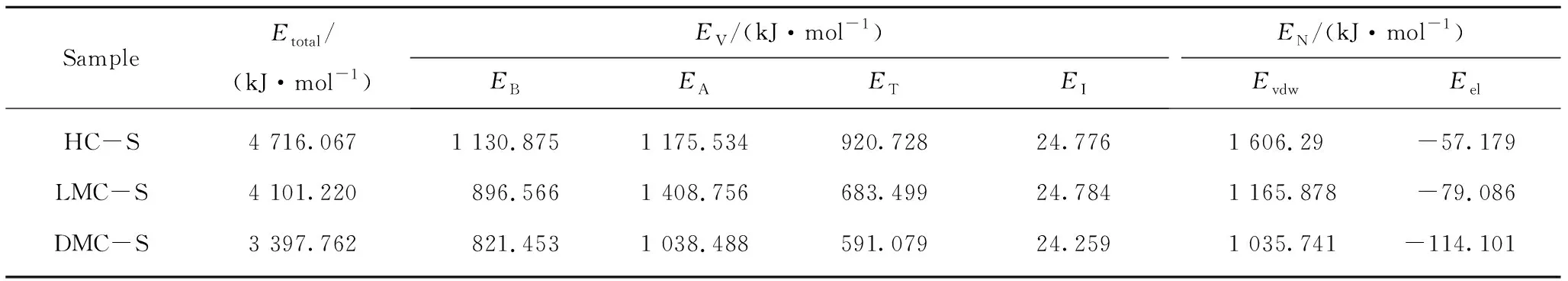
表6 族组分骨架分子最低势能构型的能量参数Table 6 Energy of group components skeletons
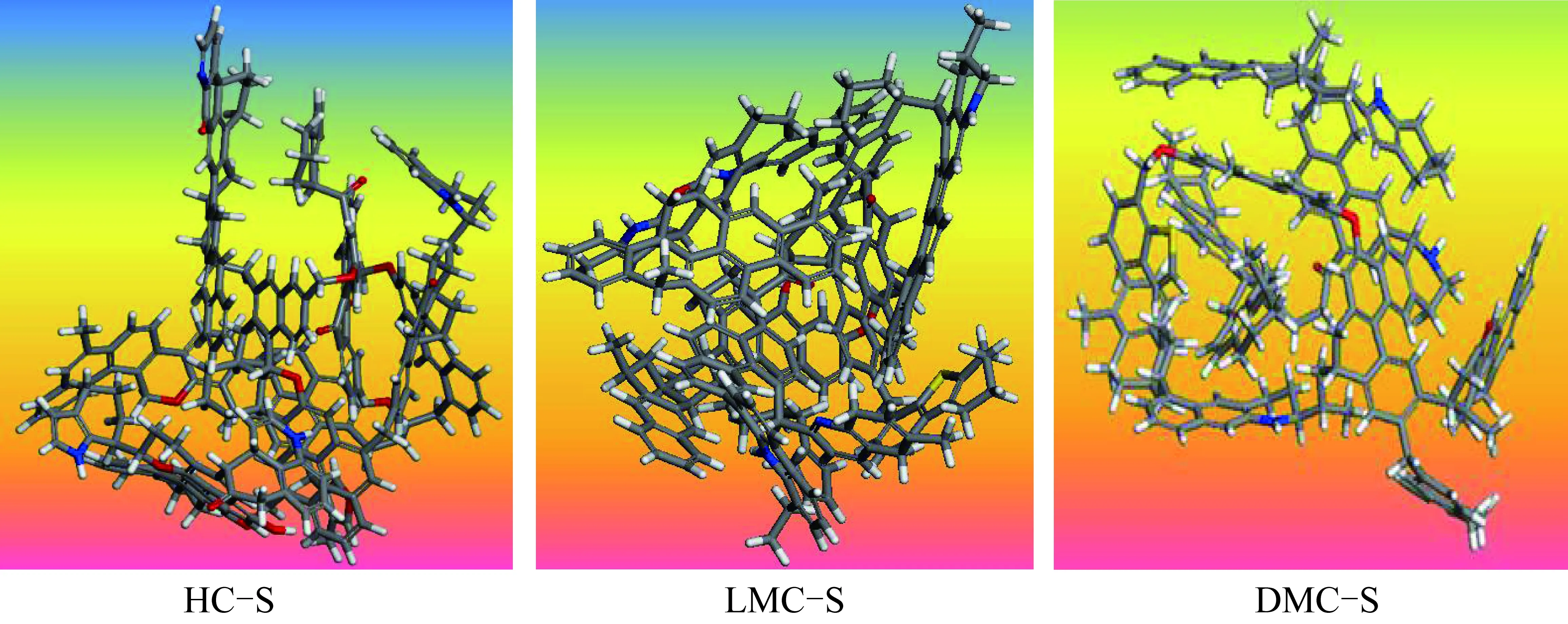
图6 族组分骨架分子最低势能构型Fig.6 Energy-minimum conformation of chemical structural models of group component skeleton
3.5 族组分骨架分子的最适密度
在分子动力学模拟中,力场选择至关重要。通过对比模拟密度与实验密度的一致性,可验证力场选择及所建分子模型的合理性。本研究所用力场下的分子势能与密度关系曲线如图7所示。根据文献[33],以曲线中第2个极小值点为最适密度点。可见, LMC-S模拟密度最大,为1.20 g/cm3;HC-S模拟密度次之,为1.14 g/cm3;DMC-S模拟密度最小,为1.05 g/cm3,与实验结果(LMC-S实验密度为1.48 g/cm3;HC-S实验密度为0.90 g/cm3;DMC-S实验密度为1.15 g/cm3)有一定的一致性。
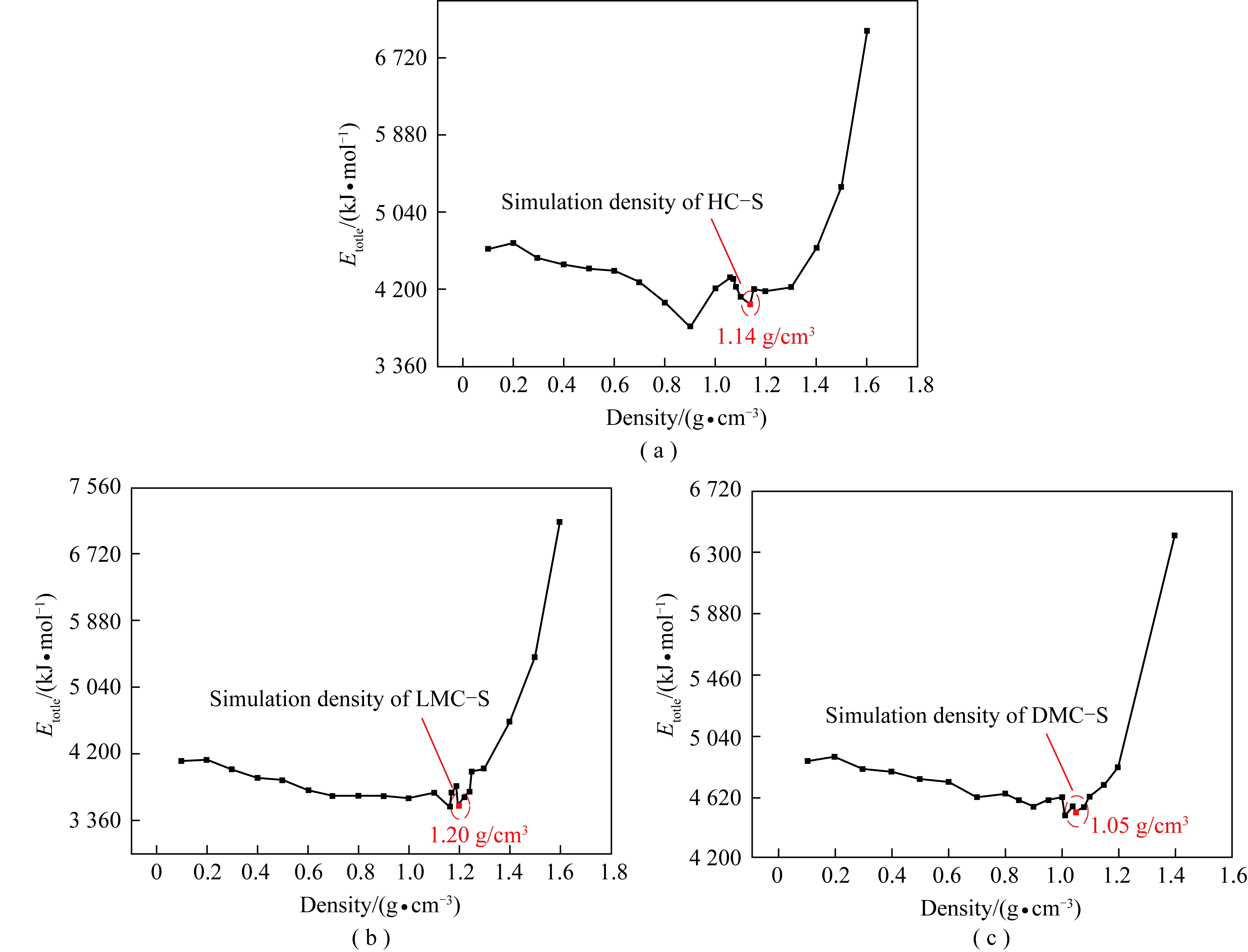
图7 族组分骨架分子势能与模拟密度的关系曲线Fig.7 Relationship between the total potential energy and the calculated densities of group component skeleton
模拟密度下各族组分骨架分子结构模型如图8所示。
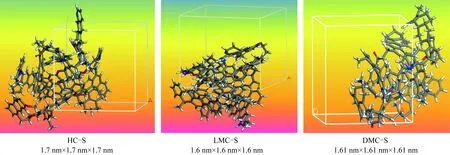
图8 相应模拟密度下各族组分骨架分子的结构模型Fig.8 Structural models of skeleton molecules of each group component with different simulated densities
3.6 族组分骨架分子不同配置单元的动力学模拟
分子间作用能的大小直接影响分子的聚集能力及聚集形态[34],在含π-体系的煤大分子聚集体中,分子间的作用主要包括芳香的π-π堆积作用、π-σ静电作用、Brønsted酸碱作用、氢键、金属配位以及烷基与烷基之间的相互作用[35]。
各族组分骨架分子构成的不同配置单元的能量参数如图9所示。可见,各族组分骨架分子形成不同个数的分子聚集体时,其能量总体呈线性增加趋势,但存在波动。总势能Etotal有不同程度波动,总键能EV波动很少,总非键能EN也有不同程度波动,即总势能的波动主要由非键能的波动引起。总键能的各项分支(键键伸缩能EB、角能EA、扭转能ET及反转能EI)也几乎不随分子个数的变化而产生波动,而非键能的各项分支(静电能Eel和范德华能Evdw)的波动情况不一。非键能的具体情况如图10所示。
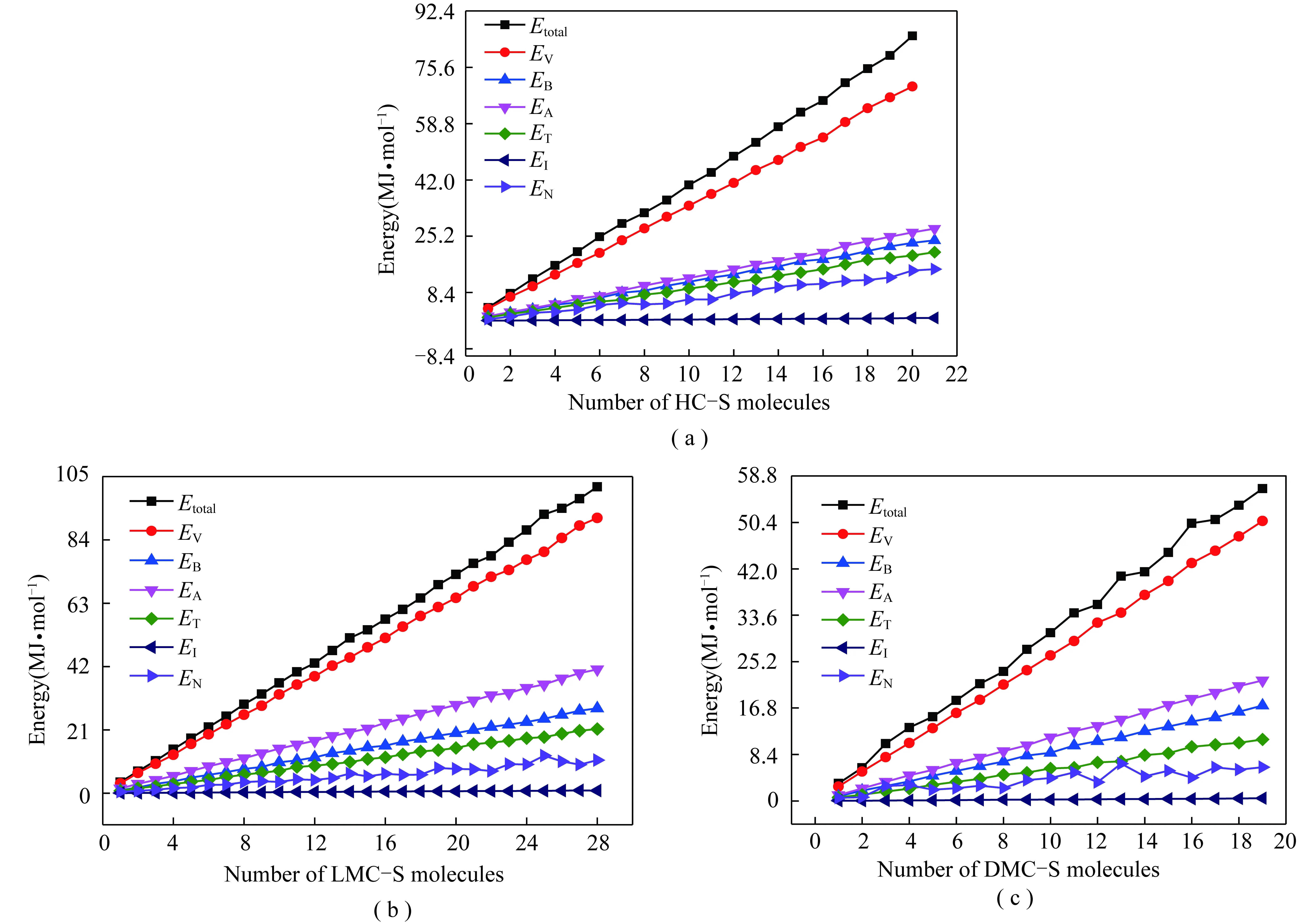
图9 族组分骨架分子不同配置单元的能量参数Fig.9 Energy parameters for different configuration unit cells of group component skeleton molecule
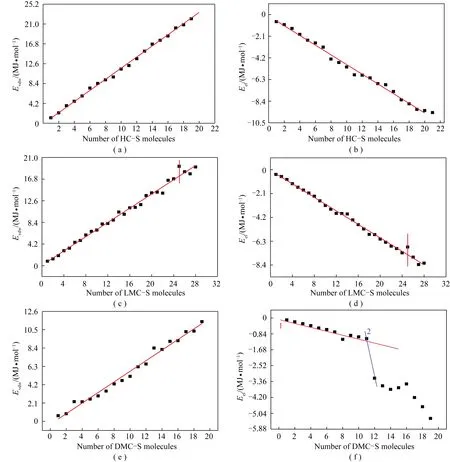
图10 各族组分骨架分子不同配置单元的静电能和范德华能变化情况Fig.10 Van der waals and electrostatic energy for different configuration unit cells of group component skeleton molecule
由图10(a),(b)可知,重质组骨架分子在形成配置单元时,范德华能和静电能的绝对值均随分子数的增加而呈线性增加,各点在直线两侧波动均较小。LMC-S分子(图10(c),(d))具有相同规律,但在分子个数为25时,其范德华能和静电能均发生明显波动,其中范德华能的绝对值增加,静电能的绝对值减小,并使总非键能(图9)增加,表明,当疏中质组以25个骨架分子形成配置单元时,可能达到稳定的聚集体状态。
DMC-S形成配置单元时,范德华能总体上也随分子数的增加而呈线性增加,但接近直线的点明显偏少(图10(e)),特别是静电能与分子个数的关系不再是完整的一条直线(图10(f)),而是分成3个区间,即分子个数为1~11,12~15和16~20的区间。除12~15区间外,另2个区间均呈明显的线性关系。特别是前2个区间交接处,即由11个骨架分子配置单元到12个骨架分子配置单元时,其静电能有非常大的能量跳跃。为了证实对密中质组中由12个骨架分子形成的配置单元(DMC-S-12)是其稳定的聚集体结构,需观察相关聚集体的结构细节。由于DMC-S分子量为2 657,其聚集体结构非常复杂。为便于观察,将其中的某个结构单元(图11中的SUN单元)提取出,如图12所示。多角度观察后发现,DMC-S-12的SUN按靠近的结构单元数量可划分成4类聚集微元,即SUN-4(指4个SUN聚集的微元,以下类推),SUN-3,SUN-2和SUN-1。除SUN-2为2个外,其余均为1个。各聚集微元具有共同的特点,即两两最接近处均为N…H 结合方式。N…H 实际上就是2个骨架分子的SUN彼此形成的分子间作用。
用同样的方法观察DMC-S-11和DMC-S-13,发现其SUN总体上处于分散状态(仅前者存在1个SUN-2),基本不能形成DMC-S-12那种较紧密的聚集微元。同时,也提取了另2组结构单元(简称SUO和SUA)(图11),结果同样不能形成图12较紧密的聚集微元,图12中数字代表该结构数量。由此可知,DMC-S-12是DMC-S聚集体的稳定结构形式,且N…H可能是形成这种稳定结构的主要因素。
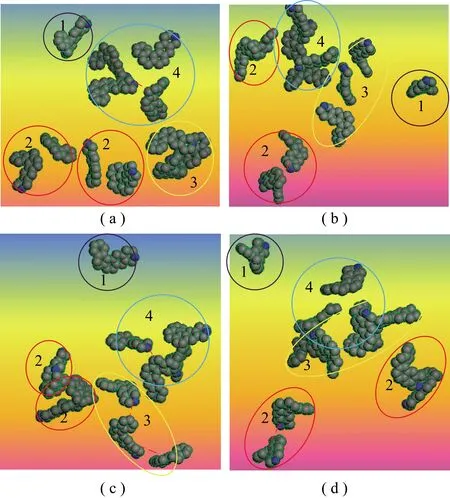
图12 DMC-S-12结构中SUN聚集微元Fig.12 SUN agregations of DMC-S-12
采用同样方式来理解疏中质组的LMC-S-25可能是其稳定的聚集体形式。由于LMC-S-25本身原子数众多且过于复杂,因此从LMC-S-25中随机提取某区域的3个骨架分子进行观察,结果如图13所示。可见该区域有3个明显的π-π构型存在。重复该工作,在其他区域也发现这种π-π构型。这说明LMC-S-25中应有较多的π-π相互作用,这可能是图9(b)中在25个骨架分子时总非键能有明显提升的原因,并且这种提升主要由范德华能上升引起(图10(c))。所以LMC-S-25可能是其稳定的聚集体形式。
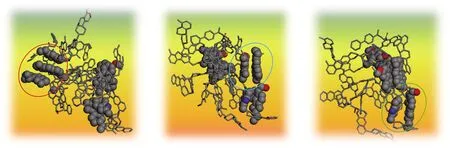
图13 LMC-S-25中随机提取的3个分子间所产生的共轭结构Fig.13 Conjugate structure of three molecules randomly selected in LMC-S-25
图10(d)中,在LMC-S-25时其静电能绝对值明显下降。FOWLER[36]在利用库伦定律统计电荷与电荷之间作用并计算2个环状体系之间的作用能时,曾得出三大规律:在面对面几何构型中以π-π排斥力占主导作用;在侧位几何构型中以π-σ吸引力占主导作用;在偏移的π-堆叠构型中也以π-σ吸引力占主导作用。前述已知LMC-S-25中有较多的π-π相互作用,且主要是面对面的几何构型,因此LMC-S-25中有更多的静电斥力,从而造成图10(d)在LMC-S-25时静电能明显下降。但这种静电斥力在数值上并不能抵消范德华能的吸引,所以总体上仍然是因LMC-S-25中的较多π-π相互作用,造成LMC-S-25成为LMC-S结构的稳定聚集体形式。
针对LMC-S-25之外的其他分子个数的聚集体,如LMC-S-24和 LMC-S-26等,通过同样的方法观察,其中的π-π相互作用极少。
对于HC-S,图9和图10都表明在计算的20个骨架分子范围内其各种能量都未显示出明显的变化,说明HC-S不会有20个骨架分子以内的稳定聚集体存在。
3.7 族组分骨架分子不同配置单元的回转半径分析
已有研究表明分子构型及分子之间的相互作用对于聚合材料的机械硬度具有非常重要的影响[37]。从概念上讲,分子的柔韧性是指给定结构承受外部扰动而变形的能力[38]。因此,分子的柔韧性在分子内相互作用过程中起关键作用[39]。回转半径可以用于描述聚合物的尺寸及分子的柔韧性大小,定义为给定构象下单体与分子质心之间距离Rg[40],公式为
式中,mi为i原子质量;ri为i原子相对分子质心的位置;N为第N个原子。
各族组分骨架分子配置单元的回转半径变化规律如图14所示。其回转半径大小分别为:LMC-S在0.95~1.13 nm,DMC-S在0.88~1.02 nm,HC-S在0.88~0.95 nm,即LMC-S回转半径最大,DMC-S次之,HC-S最小。这表明骨架分子LMC-S的柔韧性最小,形成聚集体时将更疏松;HC-S的柔韧性最大,形成聚集体时将更致密;DMC-S居中。由此, HC-S更易通过其良好的柔韧性来调整分子的空间结构而彼此聚集,从而使HC-S可能由相当多的分子来形成稳定聚集体;LMC-S因其更好的刚性(不好的柔韧性)而不易调整分子的空间结构,从而使LMC-S在聚集到25个分子时达到稳定状态(但其稳定性并不高),其后再增加分子(如26个)就很难实现更多的空间结构调整来达到稳定状态;DMC-S因其柔韧性居中,所以分子的空间结构调整难易程度也居中,其形成稳定的聚集体状态时,应有多于25个分子相聚,但由于其分子数在达到12且只在12个时,即形成了较强的N…H氢键结合,故使其形成稳定的聚集状态的分子数为12个(其稳定性强于LMC-S-25)。
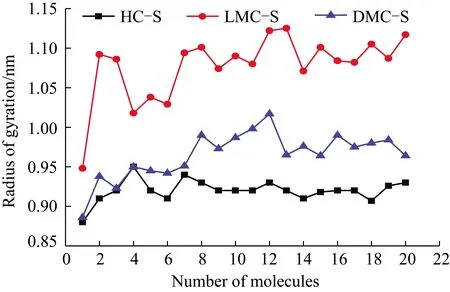
图14 族组分骨架分子配置单元的回转半径Fig.14 Gyration radius of group component skeleton molecule
3.8 族组分骨架分子形成稳定聚集体的实验验证
笔者在文献[14]中即指出,构成HC-S的大分子以芳族型结构单元为主,且骨架大分子本身即是致密的,这使得HC在所有族组分中密度最高;LMC-S主要由芳族型的中分子构成,而DMC-S含有更多的脂族型结构。这种认识是在大量的实验室实验数据基础上得出的。
(1)HC-S和LMC-S的芳族型结构,使其聚集体结构中极易形成π-π相互作用;而DMC-S的较多脂族型结构,又使其聚集体中易形成氢键相互作用。这与3.6节和3.7节的分析高度一致和互证。
(2)2008年,笔者[41]在进行族组分的TEM分析时发现,LMC主要呈现一种疏松的木耳状结构,而DMC呈现一种均匀密实的块状结构(图15)。这与3.7节讨论族组分柔韧性的分析结果互为验证。LMC-S的柔韧性小,且其稳定结构LMC-S-25中以π-π这种较弱的相互作用为主,而π-π相互作用还包含有静电斥力,使得LMC-S趋向疏松结构。特别是,LMC-S-25的这些特征,还使得LMC-S-25的聚集结构不稳定,分子间的相互作用(非共价键)易被拆散,从而导致LMC-S-25在有外作用时易于碎片化。图15(a)中大量碎片的存在,与此分析一致。DMC-S柔韧性相对较高,且其稳定结构DMC-S-12以N…H 较强氢键作用为主,因而更易于聚集并相对更致密,这是图15(b)块状均匀结构的原因。

图15 LMC和DMC固体颗粒的TEM照片[41]Fig.15 TEM images of LMC and DMC particles[41]
(3)文献[14]指出,经TEM实验观察发现,DMC(图16中黑色前景部分)实际是由5 nm的一个个纳粒聚集堆叠而成,而LMC(图16中灰白背景部分)多由50 nm以上大小不一的片状单元叠加黏连而成。前述所确定的DMC-S-12稳定结构,当其组成超胞构型时(图17),也具有5 nm的堆叠结构;而LMC-S-25的π-π弱相互作用及其静电斥力,使得LMC-S-25的分子间相互作用易被拆散而碎片化,从而形成图16(b)中大小不同的片状单元。

图16 处于CS2/NMP萃取液中时LMC和DMC的TEM 照片[14]Fig.16 TEM images of LMC and DMC in CS2/NMP extraction[14]
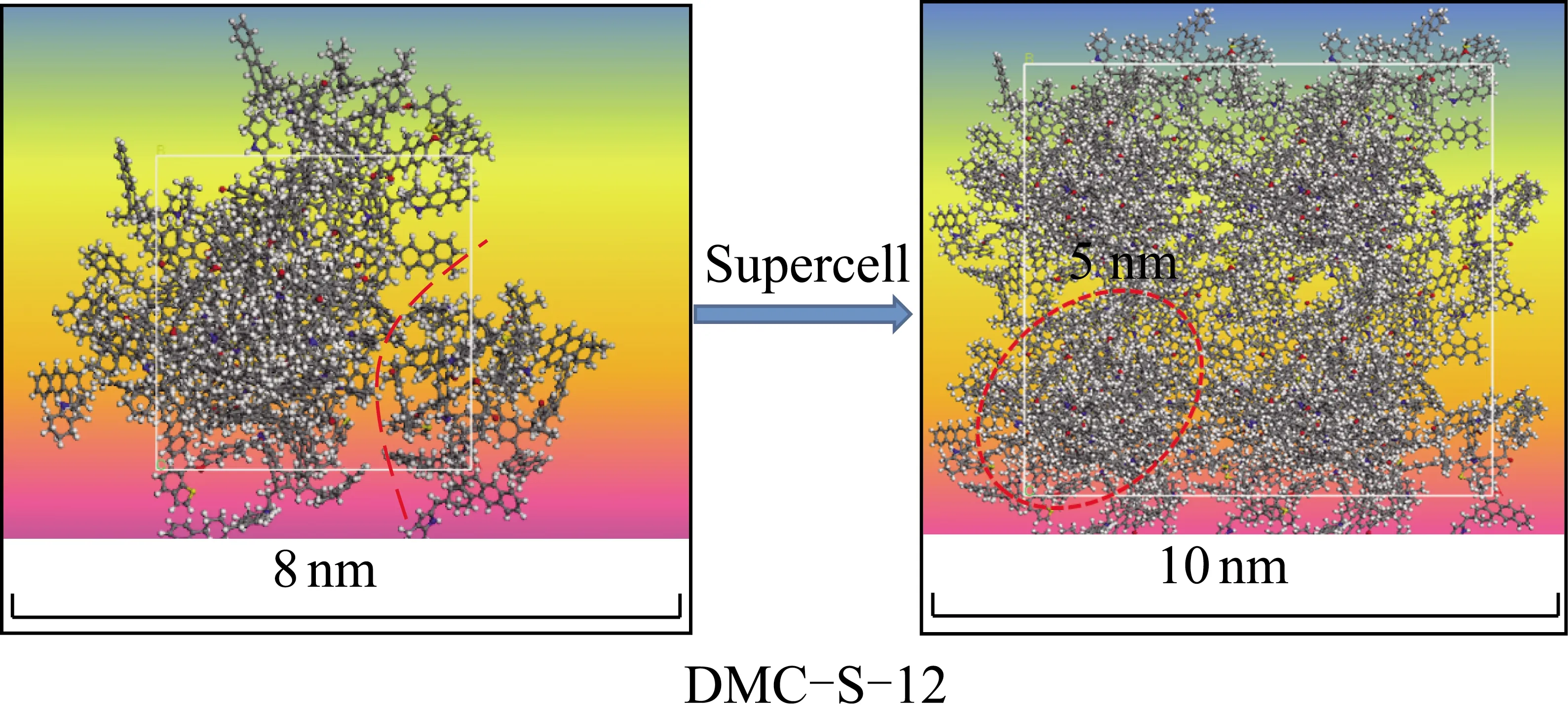
图17 DMC-S-12超胞构型Fig.17 Supercell of DMC-S-12 configuration
表7为各族组分单分子及其聚集体的模拟计算与实验结果的总结。

表7 各族组分单分子及其聚集体的模拟计算与实验结果对照Table 7 Comparison between simulation calculation and experimental results of monomers and their aggregates
4 结 论
(1)构建了煤族组分骨架的分子结构模型,其分子式:重质组HC-S为C246H186N4O15,疏中质组LMC-S为C216H183N5O5S,密中质组DMC-S为C195H179N3O4S。
(2)获得族组分骨架分子的最低势能构型及模拟密度,LMC-S模拟密度最大,为1.20 g/cm3;HC-S模拟密度次之,为1.14 g/cm3;DMC-S模拟密度最小,为1.05 g/cm3,与实验结果基本一致。
(3)当12个DMC-S聚集时,由于在不同DMC-S分子结构单元之间产生N…H相互作用而形成DMC-S-12稳定构型,其尺寸约5 nm;当25个LMC-S聚集时,该情况下产生较多的π-π相互作用形成DMC-S-25稳定构型。该机理与实验结果吻合较好。
(4)LMC-S回转半径最大,DMC-S次之,HC-S最小,表明骨架分子LMC-S的柔韧性最小,形成聚集体时将更疏松;HC-S的柔韧性最大,形成聚集体时将更致密;DMC-S居中,这与实验现象有良好吻合度。
