分子印迹-LC-MS/MS测定谷物中吡氟草胺残留量
2021-10-20王岩松贺明睿王冬妍闫晓梅罗景阳袁帅
王岩松,贺明睿*,王冬妍,闫晓梅,罗景阳,袁帅
1. 沈阳市食品药品检验所(沈阳 110124);2. 沈阳化工大学(沈阳 110142)
吡氟草胺属于类胡萝卜素生物合成抑制剂,是广谱的选择性麦田除草剂,施药后抑制类胡萝卜素的生物合成,使植物产生脱色现象,并间接地破坏光合作用,导致植物死亡[1]。吡氟草胺的毒性较强,在GB 2763—2019[2]中规定小麦中限量为0.05 mg/kg。目前,吡氟草胺的检测方法主要有气相色谱法[3]、液相色谱法[4-5]、气质联用法[6-7]和液相串联质谱法[8-10]等。液相串联质谱法具有灵敏度高、选择性强,二级质谱扫描定量可以降低假阳性的误判概率的优点。
分子印迹技术[11-17]是模仿抗原-抗体原理而人工合成的对模板分子在空间结构和结合位点形成具有“记忆功能”的高分子聚合物(MIP)的技术。MIP的特异吸附性逐渐被发掘并应用于生物传感器、化合物的提取、纯化、色谱分离等领域。分子印迹固相萃取成功地将MIP的特性融入到固相萃取的技术中,为复杂基质中目标化合物的提取、分离寻找到新的解决途径。
以吡氟草胺为模板分子,2-(三氟甲基)丙烯酸(TFMAA)为功能单体,乙二醇二甲基丙烯酸酯(EGDMA)为交联剂,偶氮二异丁腈(AIBN)为引发剂,成功制备了D-MIP,研制了吡氟草胺固相萃取小柱,应用于谷物中吡氟草胺残留量的检测。
1 试验部分
1.1 仪器与试剂
串联质谱仪(API 4000,美国应用生物系统公司);液相色谱仪(LC 20A,日本岛津公司);电子天平(BS244S,赛多利斯科学仪器有限公司);电热鼓风干燥箱(101-2,余姚市远东数控仪器厂);集热式恒温加热磁力搅拌器(DF-101S,巩义市英峪予华仪器制造厂);台式离心机(800-1,金坛区西城区新瑞仪器厂);恒温水浴振荡器(SHA-B,国华企业);电子万用炉(DL-1,北京市永光明医疗仪器有限公司);数控超声波清洗器(KQ5200DE,昆山市超声仪器有限公司);扫描电镜(FEI NOVA NanoSEM 450,FEI公司)。
吡氟草胺(95%,沈阳化工研究院);福美双(98%,沈阳化工研究院);无水乙醇(分析纯,天津市大茂化学试剂厂);TFMAA(98%,阿拉丁);AIBN(98%,麦克林);EGDMA(98%,阿拉丁);盐酸(分析纯,国药有限公司);冰乙酸(分析纯,天津市大茂化学试剂厂);甲醇、乙腈(色谱纯,美国Fisher公司)。
1.2 D-MIP的制备
准确称取0.394 g 吡氟草胺于250 mL三角瓶中,加入100 mL乙腈至完全溶解后加入一定量的TFMAA室温预聚12 h,依次加入5 mL EGDMA、0.5 mmol AIBN超声5 min,持续充入氮气10 min,封口,在60 ℃磁力搅拌24 h,取出制备的白色聚合物置于索氏提取器中,用60 mL无水乙醇-乙酸(8∶2,V/V)提取6 h,反复多次用甲醇-水(1∶1,V/V)超声洗脱至洗脱液为中性,然后用甲醇洗脱,至LC-MS/MS检测不到吡氟草胺分子为止,将聚合物置于60 ℃烘箱中烘干,得到吡氟草胺分子印迹聚合物,置于干燥器中备用。
非分子印迹聚合物(D-NIP)的制备步骤除不添加吡氟草胺,其余与D-MIP制备方法一致。
1.3 D-MIP的吸附性能测试
1.3.1 LC-MS/MS检测的条件
Symmetry C18色谱柱(150 mm×2.1 mm,3.5 μm);流动相A为水,流动相B为甲醇,分别添加0.1%的甲酸;线性梯度洗脱程序:0~1.5 min为20% B,1.5~3 min由20% B变为95% B,3~4.5 min为95% B,4.5~5 min由95% B变为20% B,5~7 min保持20% B。流速0.35 mL/min,柱温35 ℃,进样体积10 μL。
电喷雾离子源(Electron spray ionization,ESI),正离子扫描;多反应监测(Multiple reaction monitoring,MRM);电喷雾电压(Ionspray voltage,IS)5 500 V;雾化气压力65 psi(1 psi=6 894.76 Pa);气帘气压力15 psi;辅助气压力65 psi;离子源温度550 ℃;定性离子对、定量离子对、碰撞气能量(Collision energy,CE)及去簇电压(Declustering Potential,DP)见表1。

表1 吡氟草胺、福美双的质谱参数
1.3.2 吸附动力学试验
准确称取20 mg D-MIP和D-NIP,各7份,分别置于10 mL塑料离心管中,加入5 mL吡氟草胺溶液(10 mg/L)于30 ℃恒温水浴振荡。不同时间间隔同时取出1份D-MIP和D-NIP,离心后取100 μL上清液置于100 mL容量瓶中,用蒸馏水定容至刻度,取1 mL溶液过0.22 μm滤膜,供LC-MS/MS测定。以10,20,30,40,50,60和80 min时的吸附量Q与吸附时间t作图,绘制吸附动力学曲线。吸附量按式(1)计算。

式中:Q为吸附量,μg/g;V为吡氟草胺标准溶液的体积,mL;C0为吡氟草胺的初始质量浓度,ng/mL;C1为吸附后吡氟草胺的质量浓度,ng/mL;W为D-MIP或D-NIP的质量,mg。
1.3.3 静态平衡吸附试验
准确称取20 mg D-MIP和D-NIP,各6份,置于10 mL塑料离心管中,分别加入5 mL 2,4,6,8,10和12 μg/mL的吡氟草胺标准溶液,在30 ℃下恒温振荡50 min,然后离心,取100 μL上清液置于100 mL容量瓶中,用蒸馏水定容至刻度,取1 mL溶液过0.22 μm滤膜,供LC-MS/MS测定。以吸附量Q与吡氟草胺浓度C作图,绘制等温吸附曲线。
1.3.4 D-MIP选择性吸附试验
取2份20 mg D-MIP置于10 mL塑料离心管中,分别加入5 mL 10 μg/mL的吡氟草胺、福美双水溶液,恒温振荡50 min,供LC-MS/MS测定。比较MIP对2种化合物的吸附性能。
1.4 D-MIP小柱的制备及验收
1.4.1 D-MIP小柱的制备
D-MIP小柱的制备过程如图1所示。去空柱装入塞板1,加入100 mg的D-MIP,均匀覆盖在塞板1表面,装入塞板2确保聚合物厚度均匀一致后压实,塞板平面必须与柱体垂直。

图1 D-MIP小柱的装柱过程
1.4.2 D-MIP柱容量和回收率试验
D-MIP柱的活化:依次用5 mL甲醇-乙酸(9∶1,V/V)、5 mL水过柱活化。
取吡氟草胺质量浓度为1.0,2.0,5.0,10.0,15.0和20.0 μg/mL水溶液各10 mL,分别过活化后的6根D-MIP柱,用5 mL水淋洗和6 mL甲醇-乙酸(9∶1,V/V)洗脱,洗脱液稀释至10~1 000 ng/mL质量浓度范围后过0.22 μm有机滤膜,供LC-MS/MS定量分析。绘制上柱吡氟草胺标准物质的量与回收率曲线。当回收率低于80%时,上柱的量定为D-MIP净化柱的柱容量(试验在室温下操作)。
1.5 D-MIP小柱在谷物中吡氟草胺残留量检测中的应用
1.5.1 提取
试验选用大米、玉米、燕麦3种谷物样品进行试验。称取500 g谷物样品,经粉碎机粉碎,过0.850 mm(20目)孔径筛,混匀,密封,标记备用。称取10.00 g粉碎样品置于50 mL离心管中,加入5 g无水硫酸钠,再加入10 mL乙腈振荡30 min、超声5 min,以10 000 r/min离心5 min,取上清液于50 mL离心管中,残渣再用10 mL乙腈重复提取1次,合并上清液。上清液经氮气吹至近干,用5 mL水(1%甲酸)溶解待净化。
1.5.2 净化
提取液上活化后的D-MIP小柱净化,经5 mL水淋洗,6 mL甲醇-乙酸(9∶1,V/V)洗脱,洗脱液经氮气吹至近干,用1 mL甲醇定容,供LC-MS/MS定量分析。
1.5.3 方法学验证
对采用D-MIP柱净化的方法进行验证,包括方法的灵敏度、线性关系、准确度、精密度等参数。
2 结果与讨论
2.1 聚合反应条件对吸附性能的影响
2.1.1 聚合反应条件的确定
试验优化了致孔剂(乙腈)、功能单体(TFMAA)、交联剂(EGDMA)和引发剂(AIBN)的用量及配比。100 mL乙腈中加入3 mmol/L TFMAA,在室温预聚12 h,依次加入25 mmol/L EGDM和0.5 mmol/L AIBN,在60 ℃下聚合反应24 h。物料在上述比例及反应条件下,制得的聚合物产率最高,聚合物呈白色颗粒状粉末,在图2(a)中清晰可见聚合物呈空间立体网状结构。
2.1.2 模板分子与功能单体的比例对吸附性能的影响
在保持上述聚合反应条件不变的情况下,改变模板分子的加入量,制得D-MIP的SEM图。由图2可知,当模板分子加入量的比例为1∶5时,制得的聚合物团聚在一起,没有形成空间分散均匀的网状结构,不利于模板分子的洗脱,影响聚合物吸附能力。随着模板分子量的增加,聚合物的粒径逐渐减小,呈现出空间的网状结构,当模板分子的比例达到1∶3时,聚合物呈稀松的空间网状结构,颗粒表面粗糙,颗粒之间形成大量的孔隙,推测适当比例的模板分子通过静电吸附、氢键作用等在聚合物颗粒之间撑开较大的孔隙,形成大量的空间结合位点,模板分子被洗脱出去后在孔隙间留下具特异吸附性的结合位点。但是随着模板分子的量继续增加,聚合物颗粒表面变得光滑,粒径增大,孔隙明显减少,推测可能是模板分子相互作用降低了聚合物中形成孔穴的概率。因此,模板分子与功能单体的比例选择1∶3。

图2 不同比例模板分子与功能单体聚合物的SEM图片(5 μm)
2.1.3 D-MIP的吸附性能
2.1.3.1 吸附动力学试验结果
聚合物随着吸附时间的增加,吸附在聚合物表面和孔隙间的吡氟草胺分子数量增多。如图3所示:吸附时间在10~20 min之间,吸附速率最快;在20~50 min之间,吸附量以比较缓慢的速率持续增大;吸附时间增加到50 min时,吸附量达到饱和状态;继续延长吸附时间,吸附量还会出现略微的降低趋势。在吸附前期,吡氟草胺分子迅速附着在聚合物颗粒表面,占据表面的结合位点,随着时间的增加伴及振荡作用,吡氟草胺分子缓慢地进入颗粒间的孔隙,通过具有“记忆功能”的结合位点,找到适合的位置。随着时间继续延长,到50 min时,聚合物表面和颗粒间的位点全部占据,聚合物达到最大吸附量,D-MIP最大吸附量为1 350 mg/kg,D-NIP最大吸附量为320 mg/kg。因为在D-NIP表面和颗粒间缺少了这种“记忆功能”的结合位点,其吸附量明显低于D-MIP。

图3 D-MIP和D-NIP对吡氟草胺的吸附动力学曲线
如图4所示,D-NIP和D-MIP在放大倍数逐渐增大的情况下,能更清晰发现D-NIP颗粒粒径明显大于D-MIP颗粒粒径,图4(f)中可以看到D-MIP颗粒表面凸凹不平,颗粒间均匀分布大量的孔隙,正因为D-MIP颗粒表面的凸凹结构和孔隙,赋予了它良好的吸附性能和特异选择性。聚合物颗粒的结构分布情况进一步证明D-MIP吸附量明显大于D-NIP。

图4 不同分辨率D-NIP和D-MIP的SEM图片
2.1.3.2 静态吸附试验结果
保持50 min的吸附时间,验证聚合物在不同浓度吡氟草胺标准溶液中吸附量变化。如图5所示,D-MIP和D-NIP均表现出随着吸附吡氟草胺浓度的升高,吸附量变大的趋势。D-NIP的吸附曲线比较平稳,没有出现吸附量突越的现象,D-MIP结构中印迹孔隙和具有特异吸附性的识别位点与吡氟草胺浓度相匹配,吡氟草胺分子在D-MIP颗粒表面和空隙中迅速占位,当其质量浓度达到8 μg/mL时吸附行为达到终点,最大吸附量为1 350 mg/kg。X-NIP依靠自身的缝隙和疏松的结构也存在一定的吸附行为,饱和吸附量为320 mg/kg。
2.1.3.3 选择性吸附试验结果
在相同的试验条件下,比较D-MIP对吡氟草胺和福美双分子的吸附能力。结果显示:D-MIP对吡氟草胺的吸附量明显高于福美双,为1 350 mg/kg;其对福美双的吸附量为350 mg/kg。再次证明D-MIP结构中形成的“记忆”孔隙和识别位点只与吡氟草胺分子相匹配,与福美双的分子结构不具有识别功能。
2.2 D-MIP净化柱性能试验结果
净化柱的柱容量是指净化柱对一种或多种溶质可容纳的最大量。试验比较D-MIP净化柱中D-MIP粉末的装柱质量对吸附性能的影响。当装柱质量较少时,柱容量较低,在淋洗过程容易损失目标物,限制小柱的应用。D-MIP粉末的装柱质量也不宜过多,否则会导致目标物不易洗脱出来,增加甲醇的用量。综合考虑,D-MIP粉末的装柱质量选择100 mg。
试验选用吡氟草胺质量浓度为1.0,2.0,5.0,10.0,15.0和20.0 μg/mL水溶液各10 mL分别过D-MIP净化柱,经LC-MS/MS定量分析,计算不同浓度点的回收率,结果如图5所示。当吡氟草胺上柱量在100 μg以下时,回收率保持在90%以上;当上柱量超过135 μg时,回收率降低至80%以下。为保证净化柱的良好性能,确定100 μg为D-MIP净化柱的柱容量。

图5 D-NIP和D-MIP的静态吸附曲线

图6 吡氟草胺溶液过柱后的回收率
2.3 D-MIP净化柱在谷物中吡氟草胺检测方法中的应用
将分子印迹固相萃取小柱用于大米、玉米和燕麦等谷物样品中吡氟草胺残留量的检测,优化前处理条件和D-MIP净化柱的活化、淋洗和洗脱等步骤。色谱条件和质谱条件按照1.3.1节。吡氟草胺和福美双质量浓度为10 μg/L的MRM色谱图如图7所示。
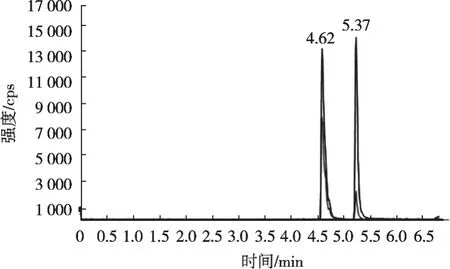
图7 吡氟草胺与福美双的MRM色谱图
由表2可知,3种基质中吡氟草胺的定量限均为0.1 μg/kg,在0.1~20 μg/kg线性范围内线性关系良好,线性关系系数r≥0.999 0。当3种基质中的添加浓度分别为0.1,0.5和5 μg/kg时,其平均回收率在89.6%~102.9%之间(n=6),平均精密度SRSD为4.41%。

表2 不同基质中吡氟草胺的相对回收率、精密度和线性关系试验结果(n=6)
3 结论
试验采用本体聚合法,乙腈为致孔剂,TFMAA为功能单体,EGDMA为交联剂,AIBN为引发剂,制备吡氟草胺的分子印迹聚合物,D-MIP的最大吸附量为1 350 mg/kg。应用D-MIP研制出分子印迹固相萃取小柱,净化柱具有较好的吸附性能,柱容量达到100 μg/100 mg。
应用D-MIP净化柱,建立谷物中吡氟草胺残留量的检测方法。该方法具有较高的灵敏度,定量限为0.1 μg/kg。当大米、玉米和燕麦基质中的添加浓度分别为0.1,0.5和5 μg/kg时,其平均回收率在89.6%~102.9%之间(n=6),平均精密度SRSD为4.41%,该方法具有较高的精密度和准确度。
