香菊片质量控制研究
2021-10-20贾文江
李 春,贾文江,李 宏,刘 峥
(陕西省商洛市药品检验所,陕西 商洛 726000)
香菊片由化香树果序(除去种子)、夏枯草、野菊花、黄芪、辛夷、防风、白芷、甘草、川芎9味中药提取制成,功能辛散祛风、清热通窍,用于治疗急、慢性鼻窦炎和慢性鼻炎。目前香菊片已收录的标准只有黄芪中主要成分黄芪甲苷的含量测定,但不能全面、准确地评价产品质量[1]。中药指纹图谱运用现代分析技术以图形的形式表达中药化学信息,具有整体性和模糊性的特点,全面呈现了中药各成分间的相互作用,高效、准确、全面[2]。本研究中采用高效液相色谱(HPLC)法测定香菊片中没食子酸、绿原酸、咖啡酸、升麻素苷、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、迷迭香酸、蒙花苷、甘草酸铵9种主要功效成分的含量,并建立了香菊片指纹图谱共有模式,对其共有峰进行指认和归属确定。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260 InfinityⅡ型高效液相色谱仪(美国Agilent公司);Sartorius SQP型电子天平(赛多利斯科学仪器<北京>有限公司,精度为0.1 mg);KQ-250DE型数控超声波清洗器(昆山市超声仪器有限公司,功率为250 W,频率为40 kHz);RS30UVE型实验室超纯水机(上海和泰仪器有限公司)。
1.2 试药
没食子酸对照品(批号为110831-201605,含量以90.8%计),咖啡酸对照品(批号为110885-201703,含量以99.7%计),绿原酸对照品(批号为110753-201817,含量以96.8%计),升麻素苷对照品(批号为111523-201712,含量以97.4%计),毛蕊异黄酮葡萄糖苷对照品(批号为111920-201606,含量以97.6%计),5-O-甲基维斯阿米醇苷对照品(批号为111523-201811,含量以97.4%计),迷迭香酸对照品(批号为111871-201706,含量以90.5%计),蒙花苷对照品(批号为111528-201710,含量以96.6%计),甘草酸铵对照品(批号为111731-201720,含量以97.7%计),辛夷对照药材(批号为121079-201205),防风对照药材(批号为120947-201810),夏枯草对照药材(批号为120993-201306),黄芪对照药材(批号为121462-201705),均购于中国食品药品检定研究院;化香树果序对照药材(广元市黎生中药材专业合作社,批号为Z190801),香菊片(批号分别为181003,191110,191201,191203,191204,191208,191209,规格为每片0.32 g),均购自陕西香菊药业集团有限公司;乙腈(色谱纯,美国Fisher公司);甲醇、磷酸(分析纯,成都市科隆化学品有限公司);超纯水(实验室自制)。
2 方法与结果
2.1 色谱条件[3-7]
色谱柱:Kromasill00-5-C18柱(250 mm×4.6 mm,5μm);流动相:乙腈(A)-0.5%磷酸水溶液(B),梯度洗脱(程序见表1);流速:0.8 mL/min;检测波长:230 nm;柱温:40℃;进样量:5μL。

表1 流动相梯度洗脱程序Tab.1 Gradient elution program of the mobile phase
2.2 溶液制备
取没食子酸、绿原酸、咖啡酸、升麻素苷、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、迷迭香酸、蒙花苷、甘草酸铵对照品各适量,精密称定,加甲醇溶解并定容,制成质量浓度分别为23.154 0,16.262 4,11.964 0,11.882 8,16.396 8,11.103 6,19.005 0,27.434 0,23.448 0μg/mL的混合对照品溶液。取7批样品,各20片,除去包衣,研细,取粉末1 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定质量,超声处理(功率为250 W,频率为40 kHz)45 min,取出,放冷,用甲醇补足减失的质量,摇匀,滤过,即得供试品溶液。取对照药材化香树果序、辛夷、防风、夏枯草、黄芪,按制备工艺煎煮2次,合并煎液,浓缩至干,加60%乙醇30 mL,过滤,低温蒸干,用甲醇5 mL溶解,滤过,取续滤液作为对照药材溶液。取除化香树果序(除去种子)、辛夷、防风、夏枯草、黄芪及其他药材,按香菊片处方工艺制备阴性样品,按供试品溶液制备方法制备阴性对照药材溶液。
2.3 指纹图谱建立及分析
2.3.1 方法学考察
精密度试验:取同一批(批号为191208)样品,依法制备供试品溶液,按2.1项下色谱条件进样测定6次,记录色谱图。结果以迷迭香酸色谱峰为参照峰,各共有峰相对保留时间的RSD均小于0.73%(n=6),相对峰面积的RSD均小于1.51%(n=6),表明仪器精密度良好。
稳定性试验:取同一批(批号为191208)样品,依法制备供试品溶液,按2.1项下色谱条件分别于0,2,4,8,12,24 h时进样测定,记录色谱图。结果以迷迭香酸色谱峰为参照峰,各共有峰相对保留时间的RSD均小于0.51%(n=6),相对峰面积的RSD均小于1.44%(n=6),表明供试品溶液在24 h内稳定。
重复性试验:取同一批(批号为191208)样品,依法制备供试品溶液,共6份,按2.1项下色谱条件进样测定,记录色谱图。结果以迷迭香酸色谱峰为参照峰,各共有峰相对保留时间的RSD均小于0.82%(n=6),相对峰面积的RSD均小于1.52%(n=6),表明方法重复性好。
2.3.2 指纹图谱建立及相似度评价
取6批样品(编号为S1-S6),按2.2项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录色谱图。详见图1。将6份指纹图谱依次导入《中药色谱指纹图谱相似度评价系统》(2012A版)软件,采用中位数法,设定时间窗口为0.1,经多点校正,生成指纹图谱共有模式,共得到18个共有峰。详见图2。以6批样品生成的指纹图谱共有模式为对照图谱,计算各谱图与共有模式的相似度。结果6批指纹图谱的相似度分别为0.965,0.973,0.976,0.964,0.969,0.977。
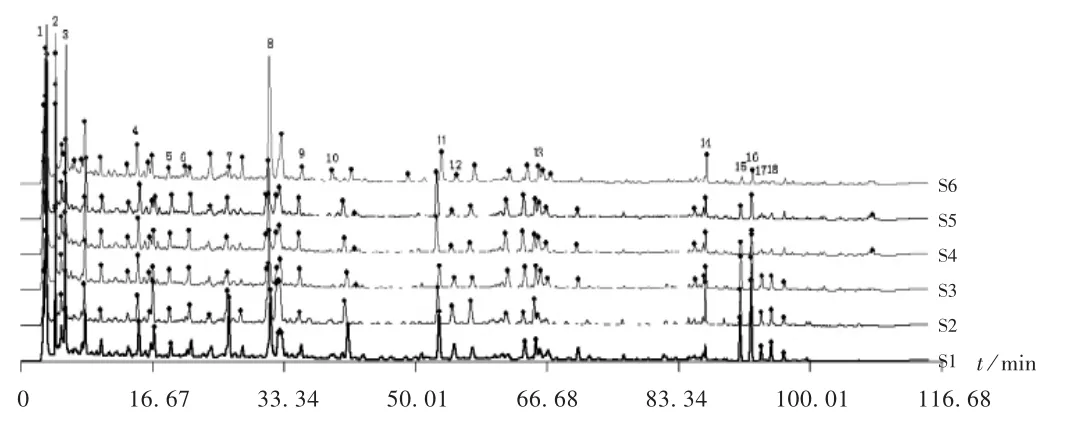
图1 样品高效液相色谱叠加指纹图谱Fig.1 HPLC superimposed fingerprints of the sample

图2 高效液相色谱指纹图谱共有模式Fig.2 The common pattern of HPLC fingerprints
2.3.3 共有峰指认与归属
取2.2项下混合对照品溶液和对照药材溶液,按2.1项下色谱条件进样测定,并与指纹图谱共有模式中各主要共有峰进行对比。以保留时间为评价指标,指认出指纹图谱共有模式中3,4,5,7,8,10,11,13,14号特征峰分别为没食子酸、绿原酸、咖啡酸、升麻素苷、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、迷迭香酸、蒙花苷、甘草酸铵;与同样条件下测得的对照药材的指纹图谱进行比较,确认1,2,3号峰来源于化香树果序,5,11号峰来源于夏枯草,7,9,10号峰来源于防风,8号峰来源于黄芪,4,6,12,15,16,17,18号峰来源于辛夷。详见图3。

图3 指纹图谱叠加图Fig.3 Superimposed fingerprints
2.4 含量测定
2.4.1 方法学考察
专属性试验:取2.2项下3种溶液各适量,按2.1项下色谱条件进样测定,记录色谱图(见图4)。结果供试品溶液色谱中,在与对照品溶液色谱相同保留时间处有相应色谱峰出现,理论板数按迷迭香酸峰(11号峰)计均大于3 000。
线性关系考察及检测限(LOD)和定量限(LOQ)确定:精密吸取没食子酸、绿原酸、咖啡酸、升麻素苷、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、迷迭香酸、蒙花苷、甘草酸铵质量浓度分别为154.360 0,65.050 0,11.964 0,11.882 8,81.984 0,11.103 6,19.005 0,27.434 0,234.480 0μg/mL的混合对照品溶液1,2,5,10,15,20 mL,分别置20 mL容量瓶中,加甲醇稀释并定容,摇匀,按2.1项下色谱条件测定,记录色谱图。以各组分对照品质量浓度(X,μg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。取2.2项下混合对照品溶液,用甲醇逐步稀释,按各组分色谱峰信噪比(S/N)分别为3∶1和10∶1时测定LOD和LOQ。结果见表2。
精密度试验:取2.2项下混合对照品溶液适量,按2.1项下色谱条件连续进样测定6次,记录峰面积。结果见表2,表明仪器精密度良好。
稳定性试验:取同一批(批号为191208)样品,按2.2项下方法制备供试品溶液,按2.1项下色谱条件分别于0,2,4,8,12,24 h时进样测定,记录峰面积。结果见表2,表明供试品溶液在24 h内稳定。
重复性试验:取同一批(批号为191208)样品,精密称定,按2.2项下方法制备供试品溶液,平行6份,按2.1项下色谱条件进样测定,记录峰面积。结果见表2,表明方法重复性良好。
加样回收试验:取已知含量的样品(批号为191208)1 g,精密称定,共6份,按2.2项下方法制备供试品溶液,分别精密加入没食子酸、绿原酸、咖啡酸、升麻素苷、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、迷迭香酸、蒙花苷、甘草酸铵对照品适量,按2.1项下色谱条件进样测定,记录峰面积,并计算各成分平均加样回收率。结果见表2。

表2 香菊片中9种成分含量测定方法学考察结果(n=6)Tab.2 Results of the methodological investigations of the content determination of the content determination of nine components in Xiangju Tablets(n=6)
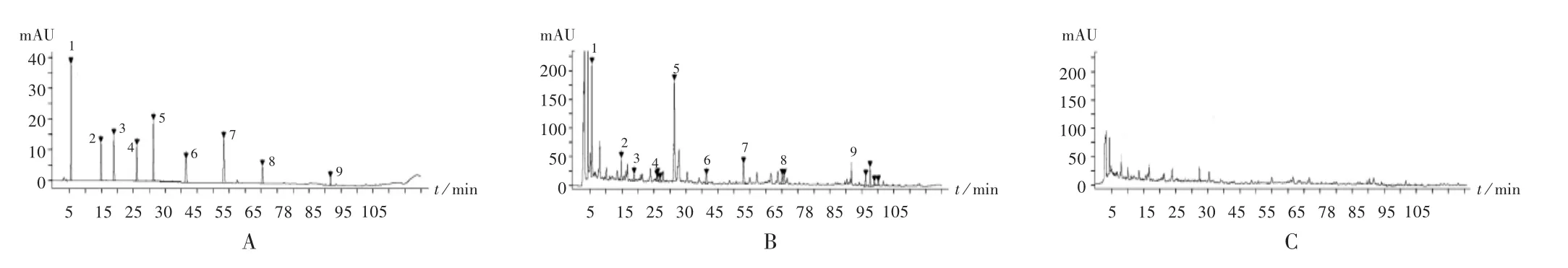
1.没食子酸 2.绿原酸 3.咖啡酸 4.升麻素苷 5.毛蕊异黄酮葡萄糖苷 6.5-O-甲基维斯阿米醇苷 7.迷迭香酸 8.蒙花苷 9.甘草酸铵A.混合对照品溶液 B.供试品溶液 C.阴性对照药材溶液图4 高效液相色谱图1.gallic acid 2.chlorogenic acid 3.caffeic acid 4.prim-O-glucosylcimifugin 5.calycosin-7-O-glucoside 6.5-O-methylvisammioside 7.rosmarinic acid 8.buddleoside 9.ammonium glycyrrhetateA.Mixed reference solution B.Test solution C.Negative reference medicinal materials solutionFig.4 HPLC chromatograms
2.4.2 样品含量测定
取7批香菊片样品各适量,依法制备供试品溶液,按2.1项下色谱条件进样测定3次,记录色谱图,按外标法分别计算各成分的含量。结果见表3。
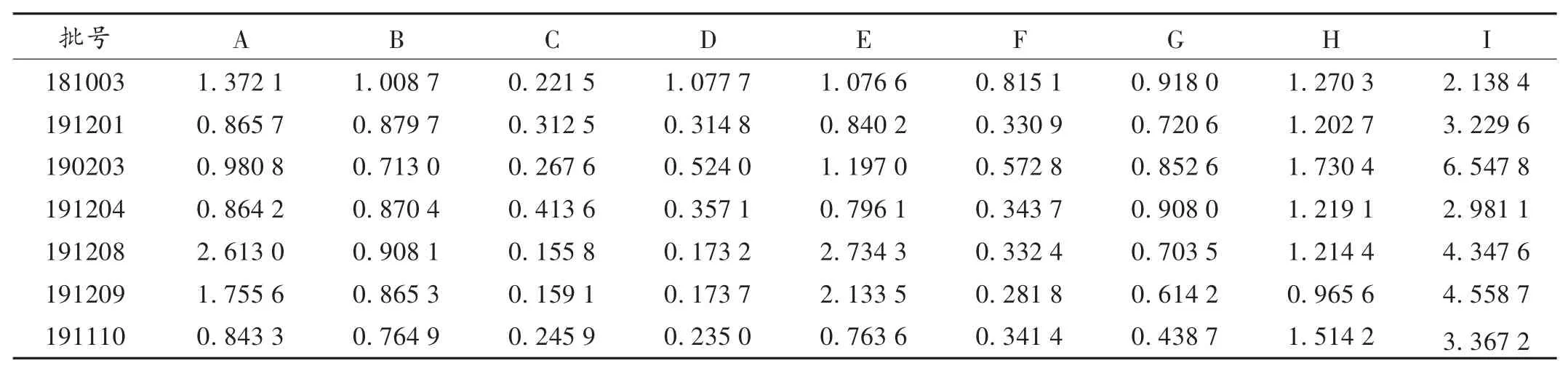
表3 香菊片中9种成分含量测定结果(mg/g,n=3)Tab.3 Determination results of nine components in Xiangju Tablets(mg/g,n=3)
3 讨论
考察了不同提取方法(超声提取和加热回流提取),不同提取时间(30,45,60 min),不同流速(0.6,0.8,1.0 mL/min),不同柱温(25,30,35,40℃),不同进样量(5,10,20μL),不同流动相体系(甲醇、乙腈、不同比例磷酸水溶液),不同检测波长(190~400 nm)对含量测定结果的影响。综合色谱峰基线平稳,保留时间适中,峰形及峰分离情况,最终选择2.1项下色谱条件。
本研究中测定了6批香菊片的指纹图谱,并采用《中药色谱指纹图谱相似度评价系统》(2012A版)建立了指纹图谱的共有模式,相似度比较结果显示,不同批次香菊片的差异较小,表明其质量较稳定。采用中位数法,经多点校正,自动匹配生成了18个共有峰,指认其中的9个指标成分,分别为没食子酸、绿原酸、咖啡酸、升麻素苷、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、迷迭香酸、蒙花苷、甘草酸铵。现代药理学研究表明,上述9种成分主要具有抗炎、抗过敏的药理学作用,与香菊片的药效相关,能较好地反映产品质量[8-11]。
综上所述,建立的方法操作简便,结果准确,重复性和稳定性均较好,可用于香菊片的质量控制,可为其选料、生产、储存、检验等过程的质量控制提供参考。
