非心脏手术患者围手术期钾离子紊乱的病理生理学及其治疗和预防的研究进展
2021-10-16石玉博宋建利刘小颖苏振波
王 宁,石玉博,宋建利,刘小颖,钱 锋,苏振波
(吉林大学中日联谊医院麻醉科,吉林 长春 130033)
钾离子(potassium,K+)是人体内最丰富的阳离子,具有维持细胞新陈代谢、保持细胞静息膜电位、调节细胞内外渗透压和酸碱平衡等功能。细胞外液K+浓度的动态平衡对神经细胞和肌肉细胞的正常功能至关重要。围手术期患者因术前胃肠道准备、禁食禁饮、并发基础疾病和服用药物等因素使得血液K+浓度的变化幅度较日常更大,急性K+紊乱发生率升高,从而导致多种意外不良事件发生,增加围手术期死亡率。研究[1-2]显示:围手术期患者K+紊乱发生率超出预期,其中住院患者低钾血症发生率约为21.0%,急诊患者低钾血症高达49.9%,胃肠手术患者低钾血症甚至可高达70.37%,即使入院常规检查正常,仍有发生严重术中K+紊乱的风险。而且术前K+异常与围手术期心血管不良事件(急性心肌缺血、心律失常和心源性猝死)发生存在关联[3],同时会增加患者死亡率,延长住院时间[4-7],须引起重视。由于心脏手术复杂程度较高且心肌细胞功能与K+息息相关,临床医生更加重视患者的K+稳态并给予积极干预,同时也进行了大量研究,如术中葡萄糖-胰岛素-钾(glucose-insulin-kalium,GIK)输注的心脏保护作用[8],术后血清K+浓度与心律关系及补K+途径研究[9-10]等相关研究,而非心脏手术患者围手术期K+稳态却未引起足够重视。
1 围手术期血清K+紊乱及其不良事件发生的临床现状
血清中K+的正常浓度为3.5~5.5 mmol·L-1,围手术期患者血清K+极易发生紊乱,普通人群中的发生率高达0.2%~16.0%,而在高危患者中发生率高达2.9%~71.0%[11]。因与术中很多原因不明的心律失常和心脏骤停存在关联,K+紊乱的症状及危害越来越受到临床重视。
1.1 低钾血症临床症状及其危害
低钾血症是临床实践中最常见的电解质异常[12],分为轻度低钾血症(3.0 mmol·L-1≤K+<3.5 mmol‧L-1)、中度低钾血症(2.5 mmol·L-1≤K+<3.0 mmol·L-1)和重度低钾血症(K+<2.5 mmol·L-1)。低钾血症症状随着低钾程度的递增而变得严重,长期重度低钾血症可引起横纹肌变性溶解,当K+<2.0 mmol·L-1时,可引起上行性瘫痪,并最终导致呼吸停止[13]。急性低钾血症(K+<3.0 mmol·L-1)可有心电图改变:起初可表现为T 波振幅降低,随后可出现ST 间期压低、T 波倒置、PR 间期延长和出现U 波,还可以引起室性心动过速/纤颤、QT 间期延长综合征、尖端扭转性心动过速和停搏等心律失常[14]。WANG等[15]报道了1 例重度低钾血症(1.13 mmol·L-1)患者心电图发生典型改变的病例,经补液补K+后好转出院。
术前访视时患者的一些临床症状有利于麻醉医生判断低钾血症的发生,如淡漠、抑郁、记忆力和定向力丧失、腹胀、呕吐、多尿、呼吸困难及心律失常等。同时患者基础状态不佳,进食困难或不能进食,需要鉴别心律失常为心脏原发还是低钾血症所致。
1.2 高钾血症临床症状及其危害
高钾血症分为轻度高钾血症(5.5 mmol·L-1<K+<6.0 mmol·L-1)、中度高钾血症(6.0 mmol‧L-1≤K+<6.5 mmol·L-1)和重度高钾血症(K+≥6.5mmol·L-1)。虽然其发生率低于低钾血症,但高钾血症人群发生率呈上升趋势且导致住院患者死亡率高达0.1%[16]。高钾血症的早期症状通常为心电图改变:起初可表现为T 波高尖,随后可出现P-R 间期延长,P 波消失,QRS 波群变平、变宽,与T 波汇合,导致连续的正弦波出现(室扑),并可进展为心室颤动或停搏;当T 波改变伴有ST 段明显抬高或压低时,提示可能为急性心肌损伤。高钾血症可以引起各种类型的心律失常,如心律不齐或传导障碍,甚至心动过缓、室性心动过速、室颤、无脉性电活动或停搏[14]。术中某些药物引起的高钾血症也可能造成严重后果,FANOUS等[17]和FLYNN等[18]曾报道因术中快速输注甘露醇引起急性高钾血症继发心脏骤停的病例。
高钾血症患者的临床表现缺乏特异性,包括神志不清、昏迷、恶心、乏力、呼吸困难、少尿和心律失常等。高钾血症的严重程度与预期的心电图结果间存在很大的脱节[19],很多严重高钾血症无典型心电图改变,这其中既有长期高钾血症心肌适应性反应降低,亦存在急性高钾血症而心肌还未发生改变,后者是围手术期最容易被忽略的,应予以足够重视。
2 K+紊乱的病理生理学病因分析
饮食摄入是人体K+的主要来源,通常每日摄入K+的含量波动范围很广,但人体总的K+含量保持相对稳定,其机制为胃肠和肾功能、酸碱状态和肾外调节维持K+摄入、排出及K+跨细胞分布的稳态,其中任一环节出现异常都会造成血清K+失衡。
2.1 K+摄入异常
人体K+摄入途径包括经肠道吸收和经静脉补充。由于肾脏保留K+能力小于排出K+,即便在低钾摄入时每天也要排K+(20~40 mmol),因此K+需要源源不断地从外界补充。
人群饮食中获得的K+浓度都远远低于推荐水平,WHO 推荐成人K+摄入量为3.51 g‧d-1,美国饮食指南[20]推荐成人K+摄入量为5.00 g·d-1,而美国成人K+实际摄入量为2.00~3.00 g·d-1[21],目前中国成人K+实际摄入量约为1.75 g·d-1[22]。临床上患者术前禁食水的时间多超过8 h 甚至更久,加之本身并发基础疾病以及入院期间紧张焦虑等各种心理因素,K+摄入量低于推荐量更多;再者,腹部手术等需肠道准备,灌肠剂导致胃肠功能紊乱,使得K+吸收障碍,同时K+排出增加,也增加了患者围手术期K+紊乱的风险。另外,中国逐渐步入老龄社会,高龄并发全身性复杂疾病伴营养状况不佳,甚至恶病质,也是低钾血症高危因素[23]。
在正常情况下,膳食K+浓度在10~400 mmol时,可通过肾脏调节全身K+浓度或血清K+浓度保持相对稳定,即使持续20 d K+摄入量为400 mmol·d-1,血清K+浓度仍保持在正常范 围内[24]。并发K+排出障碍的患者口服K+耐量下降,需要控制K+摄入量,如急性肾功能不全或慢性肾功能衰竭。发生急性胃肠炎导致K+排出过多时,需要及时补充。
静脉补充K+是中重度低钾血症的重要治疗措施,但是补充K+过多、速度过快时,极易造成医源性高血钾[13],大剂量青霉素钾盐也会造成医源性高血钾。另外术中输注大量库存血也会在短时间内引起血清K+升高,因此补K+需要全程监测。大量补液或肠外营养,忽略K+稳态时,造成稀释性低钾也是医源性低钾的常见原因。
2.2 K+排出异常
正常人体每日排出K+为1~3 g,90% 通过肾脏,10%通过肠道,少量通过汗液,另外K+的排出受下丘脑调节,夜间和清晨低,下午高,平均峰谷差为0.60 mmol‧L-1,夜间为最低点[25]。肾脏是调节K+稳态的主要器官,可在K+紊乱后数小时内通过调节K+的排泄来促进体内K+再平衡。肾脏调节K+主要通过3 个环节:肾小球滤过,近曲小管和髓袢重吸收K+,远端小管和集合管的重吸收及分泌K+。
2.2.1 肾小球滤过 血浆通过肾小球毛细血管网滤过形成原尿,滤过率正常时原尿中的K+浓度与血浆K+浓度基本相同。妊娠期,肝、肾功能受损,糖尿病肾小球硬化症患者早期肾血流量增加,肾小球毛细血管血浆胶体渗透压减小,肾小球代偿性肥大可使肾小球滤过率(glomerular filtration rate,GFR)升高,K+排泄会相应增加。
高钾血症通常发生在肾脏排泄受损时(GFR<20 mL·min-1),晚期慢性肾功能衰竭或暴发性急性肾功能衰竭,每天血清K+浓度可增加0.3~0.5 mmol·L-1;另外,肾盂或输尿管结石造成囊内压升高,少尿或无尿可以引发急性K+升高。
2.2.2 近曲小管和髓袢重吸收 原尿中90% 的K+在近曲小管和髓袢重吸收,其中约80%的K+在近曲小管重吸收,10%的K+在髓袢重吸收,而约10%的K+进入远端小管。近曲小管通过细胞旁途径重吸收一部分K+,通过跨细胞膜的水通道重吸收一部分K+。同时Na+-K+交换能够重吸收Na+,排出K+。Na+-K+与Na+-H+交换之间存在竞争性抑制,碳酸酐酶抑制剂通过抑制HCO3-重吸收使得肾小管分泌H+减少,Na+-K+交换增强,导致K+排出增加,如乙酰唑胺和噻嗪类利尿剂;髓袢升支粗段通过Na+-K+-2Cl-同向转运体将小管液中Na+、K+和Cl-同向转运入上皮细胞内,该处功能受到抑制或缺陷时,Na+、K+和Cl-重吸收减少,如袢利尿剂和Bartter 综合征。
2.2.3 远端小管和集合管的重吸收及分泌K+远曲小管和集合管是调节K+的最主要部位,能分泌K+也能重吸收K+。主细胞基底膜上的Na+-K+-ATP 酶通过泵功能实现肾小管上皮细胞内高K+、肾小管管腔中负电位状态,促进K+排出,醛固酮可以提高该酶活性,而螺内酯具有醛固酮竞争抑制作用;集合管闰细胞存在H+-K+-ATP 酶可以将H+泵入小管液同时交换K+进入细胞,实现K+的重吸收。两性霉素B 抑制集合管H+的分泌,K+重吸收受阻。
在远曲小管中表达的Na+-Cl-同向转运体(NA+-Cl--cotransporter,NCC)是调节尿液Na+和K+排泄的关键分子。高K+负荷会诱导Ca2+螯合剂被抑制,细胞内Ca2+增加,从而激活钙调神经磷酸酶,迅速使NCC 去磷酸化并促进肾脏中尿K+排泄[26]。此外,远端肾小管和集合管也存在Na+-K+交换与Na+-H+交换竞争性抑制,代谢性酸中毒引起高钾血症,高钾血症引起反常性碱性尿均与此机制有关。
远端尿流速和流量增加也会促进肾小管分泌K+,抗利尿激素作用于肾小管上皮细胞的水通道,H2O 沿着渗透梯度被动地重吸收,当远曲小管和集合管对于抗利尿激素无应答或体内缺乏抗利尿激素时尿量增加造成低钾血症,如肾性尿崩症和高渗性利尿等。
2.3 K+跨细胞分布异常
人体内K+浓度约为50 mmol·kg-1,仅2%在细胞外,K+跨细胞分布的细微变化均会影响血清K+浓度,从而造成术中急性K+浓度改变。
K+的跨细胞分布主要通过细胞膜表面的离子泵、转运蛋白、离子通道和完整的细胞膜结构功能维持。Na+-K+-ATP 酶主动摄取K+、排出Na+具有维持细胞跨膜电位的作用,该酶活性增强时促进细胞对K+的摄取,造成低钾,如胰岛素、β-肾上腺素能、茶碱和咖啡因,反之则该酶功能受抑制,导致血清K+浓度异常升高,如兴奋α-肾上腺素能受体和细胞缺氧;细胞内高浓度的K+可通过转运蛋白和离子通道顺浓度梯度流出细胞外,遗传性缺陷造成其功能异常可引起急性K+浓度改变,如周期性麻痹;H+-K+广泛存在于多种细胞的细胞膜内,体内酸碱平衡改变时影响K+稳态;细胞膜功能受损或细胞膜被破坏时也会引起K+跨细胞分布异常引起急性高钾血症,如挤压综合征和溶血。
术中诸多因素也会影响K+跨细胞分布,如麻醉药的应用,琥珀胆碱作用于神经肌肉接头的烟碱型乙酰胆碱受体引起肌细胞去极化[27],可使K+由肌细胞膜内向膜外转移导致血清K+浓度短时间内升高约0.5 mmol·L-1。而非去极化的肌松剂主要通过阻止递质到达运动终板,减少去极化的次数使K+稳定转移到细胞内。术中发生恶性高热、缺血缺氧和循环不稳定等均可以影响细胞功能,从而影响K+跨细胞分布。
2.4 其他离子与K+的相互作用
Na+是细胞外最丰富的阳离子,低钠血症会刺激肾上腺分泌醛固酮,当远曲小管和集合管对Na+的重吸收增加时,K+的分泌量同时增加。同时,部分K+通道具有Na+依赖性,细胞内Na+积累过程中K+通道的激活被认为参与了强心苷类药物导致的心律失常作用。肾小管细胞的负膜电位对从小管腔进入细胞的Na+重吸收起到重要作用。在肾小管内Na+的重吸收部位,Na+敏感的K+电导将确保膜电势维持在负电位水平,接近K+的平衡电势[28]。另外,内环境Na+和K+平衡也是维持正常细胞膜静息电位和动作电位的必要条件。
Mg2+与K+的代谢有密切关联。低镁血症可能与低钾并发,并且Mg2+浓度低可造成持续低钾血症。这是由于低镁血症使得NCC 功能下调,肾脏远端Na+排泄增加继发性引起K+排出增多,同时降低了细胞膜Na+-K+-ATP 酶活性,使得细胞不能维持正常细胞内高K+浓度[29]。Mg2+缺乏伴Mg2+依赖性或非依赖性K+缺乏的病例在普通人群中并不少见。慢性酒精中毒、心脏病、2 型糖尿病、遗传性肾性K+和Mg2+损耗(Gitelman 综合征和Bartter 综合征)、严重腹泻和呕吐及营养不良的患者在接受药物治疗时均会发生K+和Mg2+的缺乏。氨基糖苷类抗生素、顺铂、膦甲酸钠和两性霉素B通过引起Mg2+的消耗间接导致肾K+的损耗。
消化液丢失或利尿剂的使用导致Cl-丢失。Cl-偶联的Na+重吸收受到损害,增加了Na+向集合管的输送,刺激K+的分泌。在大多数危重患者中,Cl-的耗竭伴随着肾脏灌注量的减少(由细胞外容量耗竭或心、肝衰竭引起),促进醛固酮的分泌,进一步刺激K+的分泌。Cl-耗竭继发于与Cl-相关的Na+重吸收的遗传异常(Gitelman 综合征和Bartter 综合征),导致顽固性低钾血症。
Ca2+可以拮抗高钾血症的作用来保护心脏,改善心脏的收缩性和传导性。急性高钾血症治疗时可使用10 mL 10%葡萄糖酸钙静脉注射,每3~5 min重复一次,直到QRS 波变窄为止。
3 影响血清K+的疾病
疾病状态可以改变K+代谢过程,干预K+的摄入、排出与跨细胞分布状态(表1)。肾脏是调节K+最重要的脏器,影响肾脏功能的疾病均可能影响K+。醛固酮增多症、Conn 综合征和妊娠加重高血压均可引起体内醛固酮水平高于正常,K+排泄增多。Addison 病、低肾素血症和4 型肾小管性酸中毒患者的醛固酮水平低于正常,易引起高钾血症。也有临床并不多见的遗传性醛固酮代谢障碍疾病Ⅰ型假性醛固酮减少症、戈登综合征(Ⅱ型假性醛固酮减少症)和先天性孤立性醛固酮减少症等[30]均会影响K+的代谢及平衡。围手术期更应该注意上述疾病可能引起的急性剧烈K+变化。
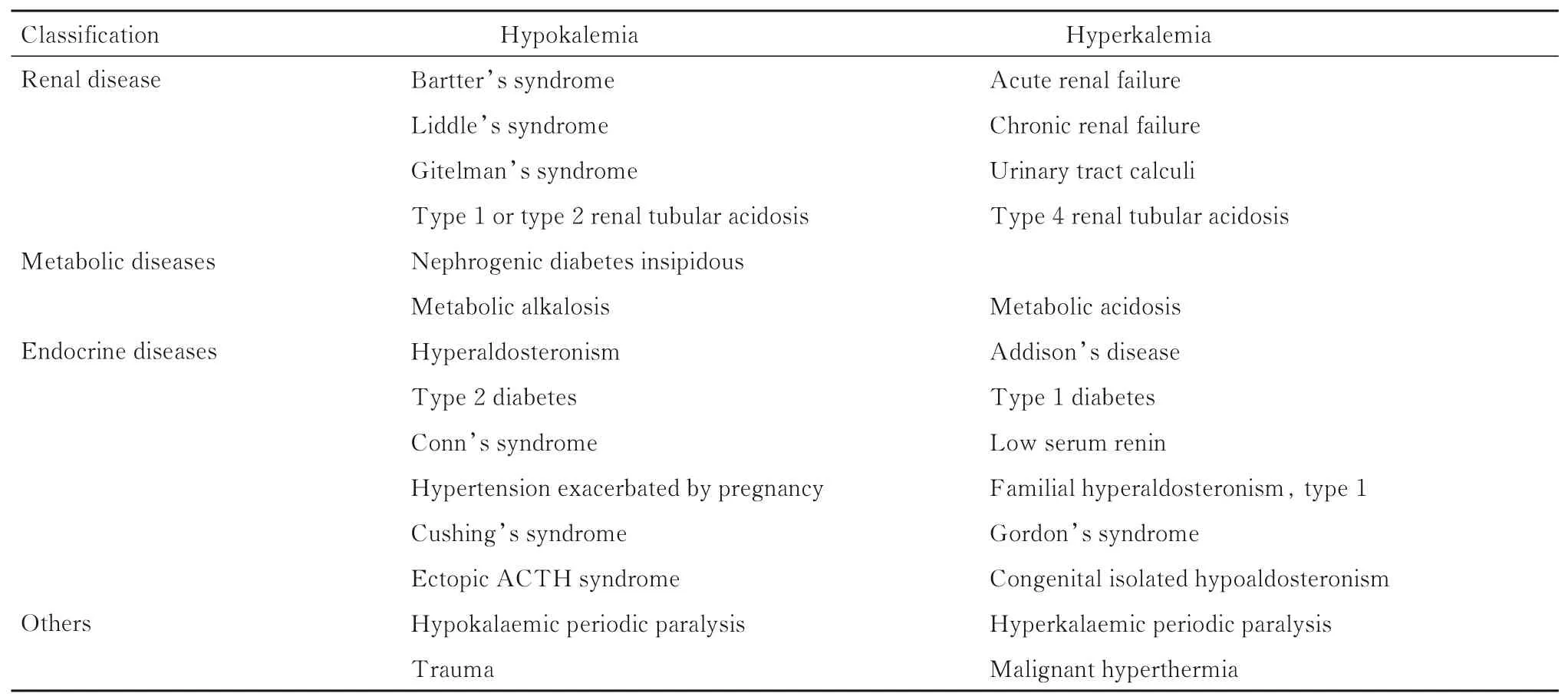
表1 影响血清K+的疾病Tab.1 Diseases affecting serum potassium
4 影响血清K+的药物
药物可以通过促进跨细胞K+转移或损害肾脏K+排泄来干扰K+平衡,同时也能增加K+的供应(表2)。肾素-血管紧张素-醛固酮系统抑制导致的肾脏K+排泄减少是已知药物导致高钾血症的最重要机制。改变跨膜K+运动的药物包括胰岛素、β 受体阻滞剂、钙通道阻滞剂、琥珀胆碱、甘露醇和洋地黄。损害肾脏K+排泄的药物主要有血管紧张素转换酶抑制剂、血管紧张素Ⅱ受体阻滞剂、直接肾素抑制剂、非甾体抗炎药、肝素及其衍生物、醛固酮拮抗剂、保K+利尿剂和青霉素及其合成衍生物。含K+制剂代表了另一组导致高钾血症的药物。提高对可诱发高钾血症药物的认识,以及监测和预防是减少与药物诱发的急性高钾血症相关的住院人数、发病率和死亡率的关键因素。
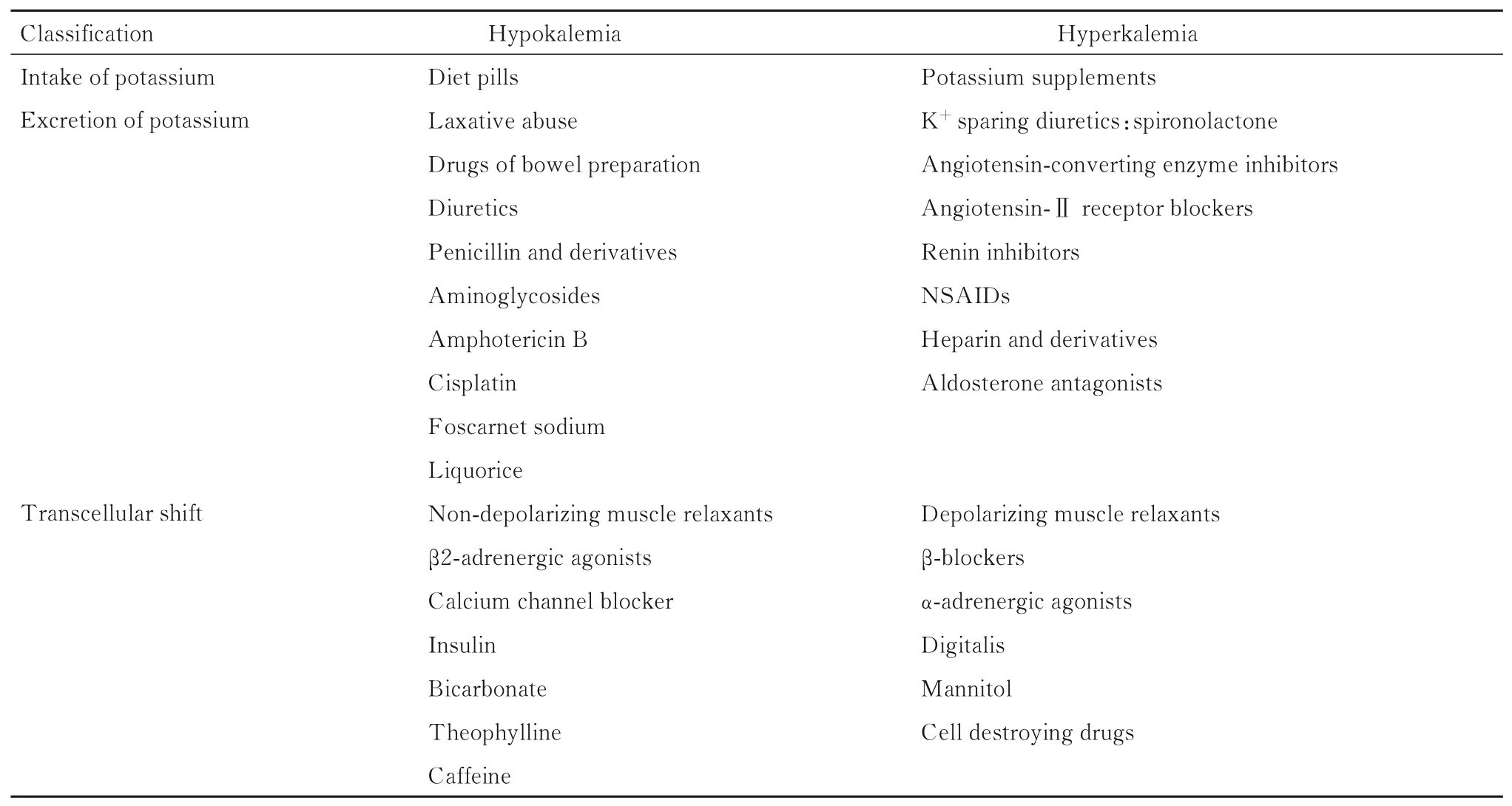
表2 影响血清K+的药物Tab.2 Drugs affecting serum K+
5 K+紊乱治疗和预防的最新进展
5.1 K+紊乱的治疗
除氯胺酮外,大部分麻醉药品均对交感神经及心功能有不同程度的抑制作用,故术中患者心肌细胞对于K+紊乱耐受力下降,K+<2.5 mmol·L-1或K+>6.5 mmol·L-1时,严重心脏损害的发生率高,必须迅速干预。临床上2.8 mmol·L-1≤K+<3.5 mmol·L-1时不必推迟手术,但是术中应该合理补充维持K+稳态,如果K+<2.8 mmol·L-1时则应视患者状态决定是否手术,伴心律失常的患者则需考虑补充K+后进行手术。但K+紊乱并不是麻醉的禁忌证,危及生命的急诊手术可在纠正K+紊乱的同时进行麻醉及手术。
轻度低钾血症患者在术前可首选口服补钾,但应注意患者有无胃肠道反应;严重低钾血症患者、胃肠道不耐受以及术中患者则需要静脉补钾,一般采用10~15 mL 10% 氯化钾注射液加入500 mL 5%~10% 葡萄糖或0.9% 生理盐水中静脉滴注。基于“先盐后糖”的原则,在血钾基本正常时改用葡萄糖溶液,预防高钾血症。补钾浓度不宜超过40 mmol·L-1,速度不宜超过13.4 mmol·h-1;当低钾伴随严重心律失常和呼吸肌麻痹等症状时,补钾量可增大,速度可加快,5%~10%葡萄糖溶液加入10%氯化钾稀释成13~67 mmol·L-1静脉滴入,速度不宜超过20 mmol·h-1。对于危及生命的严重低钾,可考虑采取非常规的补钾方式,如通过中心静脉和输液泵补钾,执行此项操作时需经验丰富的医生,并密切关注患者K+浓度变化。除此以外,研究者正在探究个体化快速补钾方式[31],先在5 min 内快速静脉给予负荷量3% KCl 溶液至血清钾浓度达3.5 mmol·L-1后,再以常规速度静脉维持,该处理方式下血清K+浓度、尿液K+浓度和细胞外液K+浓度增加速度均较传统方式更快,未来有望应用于急性致命性低钾血症患者的治疗。
高钾血症的治疗基于以下4 个关键原则:通过拮抗高钾血症的作用来保护心脏;将K+移入细胞内;促进K+排出;监测血清K+浓度预防高钾血症的复发。Ca2+可用于改善心脏的收缩性和传导性。葡萄糖、胰岛素、碳酸氢盐和β-肾上腺素能激动剂会促进K+向细胞内转运。离子交换树脂(聚苯乙烯磺酸钠和聚苯乙烯硫酸钠)会增加胃肠道的K+排泄。当在无其他有效措施时,则可考虑进行透析治疗高钾血症。帕特罗默(patiromer)和环硅酸锆钠(ZS-9)[32]是治疗急性和慢性高钾血症的新药。帕特罗默口服混悬剂的活性部分是一种不可吸收的聚合物,其在整个胃肠道均可与K+结合,但主要作用于游离K+浓度最高的远端结肠,最终增加分泌、降低血清K+浓度[33]。ZS-9 是一种选择性很高的无机阳离子交换剂,可以将K+捕获到肠道中,其K+结合容量是聚苯乙烯硫酸钠的9.3 倍,对K+的选择性是聚苯乙烯硫酸钠的125 倍以上[34]。帕特罗默和ZS-9 可能在数小时内迅速降低血清K+浓度,提示胃肠道在K+浓度调节中可能发挥更强大的作用。
5.2 K+紊乱的预防
低钾血症和高钾血症是围手术期患者易被忽视的电解质异常,因其早期症状的非特异性或存在隐匿性疾病易被其他疾病所掩盖,术中电极位置并不标准,提高了K+紊乱的诊断难度,但是心电图动态监测对于发现K+紊乱仍具有重要意义。发生K+紊乱时临床医生将更多的关注度放在对症治疗,错过干预最佳时机,造成不良后果,而早期干预可避免高钾血症和低钾血症所导致的严重不良事件。LU 等[35]证明了以入院前为起点的低血钾的预防和控制系统更加有效,同时监测血清K+浓度可以尽早纠正开腹手术患者的低钾血症,还可以提高床位利用率,因此围手术期K+紊乱的预防意义重大。
K+紊乱可能预测疾病的进展,最新研究[36-37]表明:85% 的重症新型冠状病毒肺炎(corona virus disease-2019,COVID-19)患者并发低钾血症,低钾血症是有创机械通气需求量的独立预测因素,其严重程度与COVID-19 患者症状的严重程度呈正相关关系,是COVID-19 患者严重进展的敏感生物标志物。与正常血清K+浓度患者比较,低钾血症患者在入院时空腹血糖和GCS 评分更低,症状起病至治疗时间更长,是脑卒中、心血管和肾脏不良结局的预测因子[38]。最近的一项回顾性研究[39]显示:血清K+浓度5.5~6.0 mmol·L-1与4 d内的住院风险增加存在相关关系,血清K+浓度升高(6.0~6.5 mmol·L-1或更高)与死亡和急诊就诊存在相关关系。因此,临床监测血清K+浓度更加必要。
目前常用中心实验室静脉血清K+测定和动脉血气(arterial blood gases,ABG)血浆K+测定监测K+浓度。中心实验室静脉血清K+测定平均耗时为60 min,当样本溶血、脂血、样本不足甚至样本丢失或设备处于校准时耗时会更长。ABG 行K+浓度测定耗时仅为2 min,在急诊、手术间和ICU 具有应用优势。因此ABG 的K+浓度测定应用越来越广泛,研究[40-41]表明虽然ABG 血浆测定K+浓度与中心实验室静脉血清K+浓度之间差异有统计学意义,但偏差未超过美国临床实验室改进修正案确定的限值。因此,ABG 测量的K+浓度是可靠的,具有临床意义。
K+紊乱的一级预防是为血清K+紊乱高危患者监测血清生化指标,并停用影响血清K+浓度的药物,同时提倡口服含K+的糖溶液。加速康复外科(enhanced recovery after surgery,ERAS)理念已经由推荐围手术期患者术前饮用碳水化合物转变为推荐术前饮用加入电解质的含糖饮料。KAMOLPORNWIJIT 等[42]证明了口服氯化钾可预防妇科腹腔镜手术前肠准备的低钾血症。二级预防重要的是在初次治疗K+紊乱后监测血清K+浓度,并通过治疗原发致病因素来预防K+紊乱的复发,有充血性心力衰竭或心肌梗死病史的患者应维持4.0~4.5 mmol·L-1的血清K+浓度[43]。
5.3 与K+紊乱治疗相关的不良事件
高浓度K+容易造成外周血管静脉炎;补充K+量过多、速度过快易造成医源性高血钾[13];地高辛诱导的高血钾症患者,急性输注葡萄糖酸钙会导致心脏传导阻滞[44];胰岛素和沙丁胺醇的使用可以导致低血糖和心动过速;外源性盐皮质激素的应用可增加容量超负荷并导致严重的心肺并发症;大量静脉输注NaHCO3可能导致急性高渗,引起桥脑中央髓鞘溶解症[45];输注NaHCO3也存在发生急性肺水肿和急性心肌梗死,增加心脏手术患者死亡的风险[46];利尿剂可引起血容量减少、Na+代谢障碍、低镁血症、肾结石和痛风发作;聚苯乙烯磺酸钠与许多肠道坏死病例有关[47];帕特罗默已被证明在高剂量时会导致低镁血症、高钙尿,甚至水肿[48];急性透析通路的中心置管易导致大量围手术期并发症和中心静脉的损伤,低钾透析液(<2.0 mEq·L-1)被用于治疗严重的高钾血症,但细胞外K+浓度迅速(在几分钟内)降低的影响还有待进一步研究。
综上所述,众多严重临床不良事件与围手术期K+关系密切[3-4,7,13,17-18],临床医生应该更加关注非心脏手术患者围手术期K+浓度的变化,提高对K+紊乱的预防意识,尤其是对使用影响K+稳态药物及并发影响K+稳态疾病的患者应该监测K+浓度,对出现临床症状的K+紊乱应该积极治疗,并且尽量避免不良事件的发生。同时提高对麻醉诱导药物可能引起的急性K+紊乱的警惕性。此外,轻度低钾或血清K+浓度处于正常值下限但总K+浓度下降的患者是否应该预防性补充K+也是研究方向之一。
